

Fluorine containing chalcones as anticancer agents: Solvent-free synthesis and cytotoxicity against HepG2 cancer cells
- Faculty of Chemical Technology, Ho Chi Minh City University of Technology and Education, Ho Chi Minh City, Vietnam
- Faculty of Medicine and Pharmacy, Thanh Dong University, Hai Duong Province, Vietnam
- Institute of Applied Materials Science, Academy of Science and Technology, Ho Chi Minh City, Vietnam
Abstract
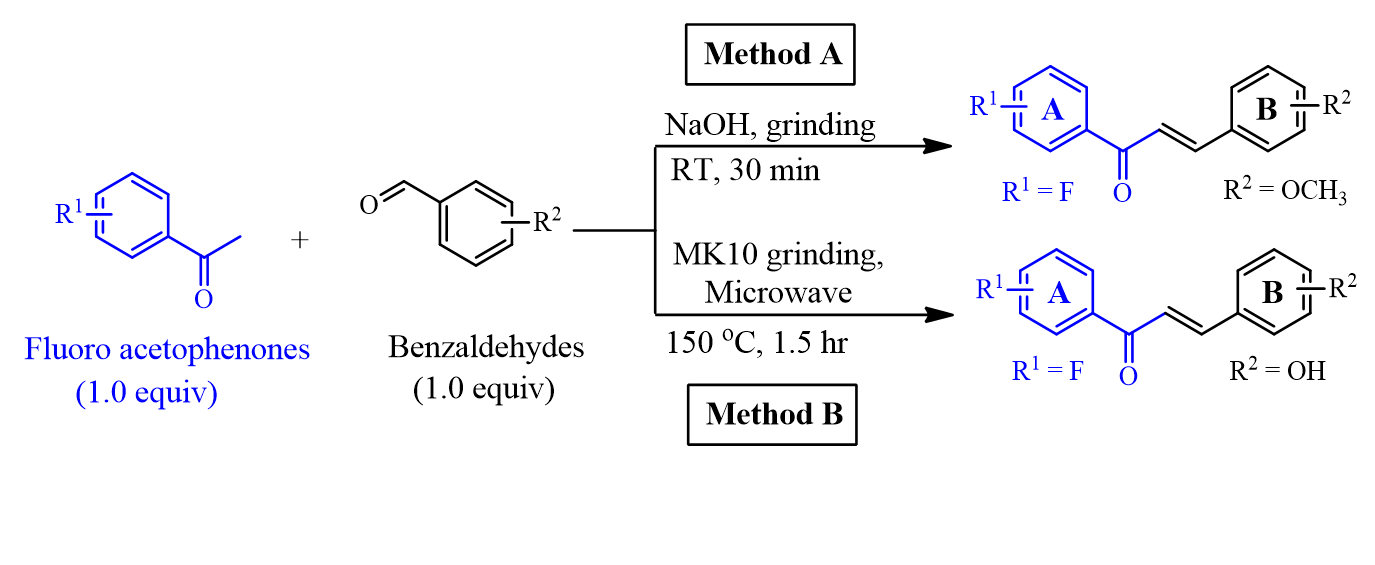
Introduction: Chalcone analogs are commonly synthesized by the base-catalyzed Claisen-Schmidt condensation reaction of acetophenones and aromatic benzaldehydes. The shortcomings of the traditional protocols are side-products. Grinding and microwave-assisted methods under solvent-free conditions are green approaches for the preparation of chalcones. Herein, we focused on the synthesis of chalcone-based analogs bearing –F/-OCH3/-OH groups on both A and B rings using grinding and microwave irradiation procedures. In our efforts to search for anticancer agents, fluoro-containing chalcones were subsequently assayed for cytotoxic activities against HepG2 cells.
Method: The fluorinated chalcones were synthesized using cross-aldol reactions between fluoro acetophenones and benzaldehyde analogs. Fluorinated chalcones bearing –OCH3 at positions of the ring B were prepared by grinding reagents with solid NaOH under solvent-free condition, while fluoro-containing chalcones bearing –OH substituents in the B ring were obtained from the clay-catalyzed microwave irradiation in the absence of solvent. Chemical structures of fluorinated chalcones were elucidated by NMR and MS spectra. The inhibitory activity against HepG2 cells was evaluated through the half-maximal inhibitory concentration (IC50, µM).
Results: Twelve fluorinated chalcones (1a-1c, 2a-2c, 3a-3c, 4a-4c) were successfully synthesized in 33-49% yields. Compounds 3a, 3b, 3c and 4c are novel fluoro-containing chalcones. Except for compounds 1a-1c, fluorinated chalconoids exhibited cytotoxic activities against HepG2 cancer cells with IC50 values in the range 67.51-108.20 µM. Chalconoid 2a showed a highest inhibitory activity with an IC50 value of 67.51±2.26 µM.
Conclusions: The grinding of reagents together prior to adding solid NaOH resulted in desired products. The clay-catalyzed microwave irradiation in the absence of solvent is effective to prepare hydroxy chalcones. Among synthesized structures by eco-friendly approaches, four fluorinated chalcones 3a-3c and 4c are novel compounds. The scaffold of 4¢-F-3-OCH3 might contribute to the cytotoxicity against HepG2 cells.
INTRODUCTION
Chalconoids are a common group of natural products. Chemically, chalcones are composed of two aromatic rings attached at the two ends of a three-carbon α,β-unsaturated carbonyl linker1. In view of this, chalconoids have been commonly synthesized from aryl methyl ketones with benzaldehydes via a base-catalyzed Claisen-Schmidt condensation reaction in the liquid phase for several hours2, 3. However, this procedure has several drawbacks, such as long reaction times with probabilities of byproducts (Cannizaro and/or Michael addition products)3, 4. To limit disadvantages, the condensation of acetophenones and aromatic benzaldehydes was carried out in the solid phase in good yields5, 6, 7. A series of chalconoids were previously prepared via solvent-free grinding and microwave-assisted methods6, 7, 8.
From a biological point of view, chalcones exhibit diverse properties, including antioxidant9, anti-inflammatory10, antidiabetic11 and anticancer12 activities. In particular, the different functional groups on both aromatic rings of chalcones are responsible for their anticancer properties13. Interestingly, the substitution of hydrogen with fluorine in chalcone molecules has improved anticancer activity14, 15. The potent activities can be attributed to the stereochemical changes in the C‒F bond, which modulate the reactivity and stability of overall molecules in metabolic transformations in biological systems16, 17. The antiproliferative activities of fluorinated chalcones against HepG2 cells have been reported. Considering their cytotoxicity against HepG2 cells, Burmaoglu et al. reported that fluoro-substituted chalcones were more effective than nonfluorinated compounds were18. The bioisosteric substitution of fluorine instead of hydrogen resulted in increased activities of fluoro-containing chalcones. The in vitro and in silico studies on fluoro-substituted chalcones indicated that the anticancer properties of fluorinated chalcones toward HepG2 cells are dependent on the position, number and size of the substituents. The methoxy (-OCH) and fluoro (-F) groups contributed to the potent anticancer activity19.

General synthetic procedures for producing fluorinated chalcones by grinding at room temperature (RT) (Method A) and microwave irradiation at 150 °C with montmorillonite K10 (MK10) clay as a catalyst (Method B).
Motivated by these findings, we synthesized chalcones with the -F group at various positions of ring A and –OCH/-OH at positions of ring B via microwave-assisted, MK10-catalyzed and grinding procedures under solvent-free conditions. In continuation of our studies on anticancer agents, we evaluated their cytotoxicity against HepG2 cells.
MATERIALS AND METHODS
Chemicals
All chemicals and solvents used for synthesis and purification were purchased from suppliers without further purification. 4'-Fluoroacetophenone (99%, Acros Organics), 3'-fluoroacetophenone (99%, Acros Organics), 3',4'-difluoroacetophenone (96%, Fisher), 4-methoxybenzaldehyde (98%, Acros Organics), 3-methoxybenzaldehyde (99%, Acros Organics), 2,3-dihydroxybenzaldehyde (98%, Acros Organics), and 4-hydroxybenzaldehyde (98%, Acros Organics) were selected as starting materials for the synthesis of fluorinated chalconoids in the present work. Sodium hydroxide (NaOH, 99%, China) and montmorillonite K10 (surface area 220–270 m/g, Sigma‒Aldrich) were used as catalysts for grinding and microwave-assisted methods. n-Hexane (HEX, > 99%, J. T. Baker), dichloromethane (DCM, > 99%, J. T. Baker) and ethyl acetate (EA, > 99%, J. T. Baker) as solvents were stored in 4.0 L glass bottles.
Analytical methods
Nuclear magnetic resonance-NMR spectra were recorded on a Bruker Avance (600 MHz (H), 150 MHz (C)). Chemical shifts δ [ppm] were referenced to residual protonated solvent signals of: CDCl: δ = 7.26 ppm (H); 77.16 ppm (C), DMSO-d: δ = 2.50 ppm (H); 39.52 ppm (C). The proton and carbon signals are abbreviated as s (singlet), brs (broad singlet), d (doublet), dd (doublet of doublet), td (triplet of doublet), t (triplet), and m (multiplet). Mass spectrometry-MS measurements were performed on an AGILENT 1200 series LC‒MSD. The intermediates and products were monitored and purified via thin layer chromatography-TLC (silica gel 60 F, Merck) and flash column chromatography-CC (silica gel 0.035–0.070 mm, Merck). UV–detection was carried out at λ = 254 and 365 nm to visualize spots via TLC.
Synthesis of fluorinated chalcones (1a-1c; 2a-2c) via a solvent-free grinding method (Method A)
A mixture including benzaldehydes (5.0 mmol, 1.0 equiv) and fluorinated methyl ketones (5.0 mmol, 1.0 equiv) was ground in an open mortar for 10 min at room temperature using a pestle. Sodium hydroxide (5.0 mmol, 1.0 equiv.) catalyst was added sequentially, and the mixture was ground for 20 min. The progress of the synthetic reactions was monitored by TLC. The reaction mixture was then added to distilled water (15 mL) and transferred completely to a separatory funnel (125 mL). The crude product was extracted with dichloromethane (DCM, 3x15 mL). The combined organic layer was separated, dried over NaSO and evaporated via a rotary evaporator. The residue was chromatographed by CC over silica gel and eluted successively with n-hexane (HEX)/ethyl acetate (EtOAc) (98/290/10, v/v) to afford fluorinated chalcones 1a-1c; 2a-2c.
Synthesis of fluorinated chalcones (3a-3c; 4a-4c) via a solvent-free, MK10 grinding, microwave-assisted method (Method B)
Starting materials of hydroxy-substituted benzaldehydes (5.0 mmol, 1.0 equiv) and fluorinated acetophenones (5.0 mmol, 1.0 equiv) and montmorillonite K10 clay catalyst (1.0 g, 0.2 g/mmol) were first ground using an open mortar with a pestle at room temperature for 20 min. Consequently, the mixture was transferred to a 10 mL vial sealed with a 10 mL cap and heated at 150 C under microwave irradiation (Microwave Reactor Discover 2.0, CEM, USA). The “standard” method (vessel type: Pyrex; control type: standard; temperature: 150 C; time: 1.5 h) was selected to perform the reactions. After irradiation for 1.5 h, the mixture was cooled and diluted with 45 mL EtOAc. The solution was filtered through a Büchner funnel to remove the catalyst. The organic phase was dried over NaSO and filtered, and the solvent was removed under reduced pressure. The crude product was purified via CC (eluent: HEX/EtOAc = 95/5→ 80/20).
Assays for HepG2 cells
Fluorinated chalcones were assayed for their cytotoxic activities against human hepatocellular carcinoma (HepG2) cells in vitro via the modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma‒Aldrich, USA) method20. Doxorubicin (0.01 mM) was used as a positive control to assess validity. The HepG2 (ATCC, HB-8065TM, USA) cell line was seeded in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and other components. The cells (3×10) were grown in a 37 C humidified incubator with 5% CO. Fluorinated chalconoids were dissolved in 10% dimethylsulfoxide in medium (DMSO, Aldrich 99.9%) at a concentration of 20 mg/mL. The final concentrations of the samples in the wells were 128, 32, 8, and 2 µg/mL. Each concentration was tested in four separate wells per 96-well plate and in duplicate (4×2). The concentration of DMSO in the test wells was 0.5%. The growing cells were inoculated at the appropriate concentration (190 µL volume) into each well of a 96-well plate. Sample solutions were applied (10 µL volume) to the culture wells, and the cultures were incubated for 72 hours at 37 C. MTT was prepared at 5 mg/mL, and 10 µL was added to the microculture wells. After 4 hours of incubation at 37 C, 210 µL of culture medium was removed from each well, and the formazan product was dissolved in 100 µL of DMSO. The optical density (OD) at 540 nm was measured with a BioTek microplate reader. The percent inhibition I (%) of HepG2 inhibitory activity was calculated via the following equation:
I (%) = [(OD – OD)/(OD - OD)] × 100%
The half-maximal inhibitory concentration (IC) was determined from the I% values at various inhibitor concentrations.
RESULTS
Solvent-free synthesis of fluorinated chalconoids
Among the twelve fluorinated compounds synthesized in our present work, four fluorinated chalcones, 3a, 3b, 3c and 4c, were synthesized for the first time. The commercial fluoro-substituted acetophenones were coupled with benzaldehydes via a grinding method under solvent-free and NaOH-catalyzed conditions to afford chalcones 1a-1c and 2a-2c (Figure 2) in 41-49% yields (

Chemical structures of fluorinated chalconoids.
In addition, with the MK10 catalyst in hand, we applied the solvent-free, microwave-assisted method for the synthesis of chalcones bearing hydroxy groups in ring B. After 1.5 h of irradiation at 150 C, work-up followed by CC resulted in the purification of 3a-3c; 4a-4c in 33-44% yields (Figure 2 and
(E)-1-(4'-Fluorophenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (1a): Yield 42%, white solid, CHFO [256.09 g/mol]; R = 0.53 (HEX/EtOAc = 95/5); m.p. 104.7 °C; H-NMR (600 MHz, CDCl): δ = 3.86 (s, 3 H, -OCH), 6.94 (d, J = 9.0 Hz, 2 H, H3,5), 7.17 (dd, J = 9.0 Hz, J = 8.4 Hz, 2 H, H3',5'), 7.37 (d, J = 15.6 Hz, 1 H, Hα), 7.60 (d, J = 8.4 Hz, 2 H, H2,6), 7.78 (d, J = 15.6 Hz, 1 H, H), 8.04 (dd, J = 9.0 Hz, J = 5.4 Hz, 2 H, H2',6') ppm C-NMR (150 MHz, CDCl): d = 55.5 (-OCH), 114.5 (C3,5), 115.6 (d, J = 21.0 Hz, C3',5'), 119.4 (Cα), 127.6 (C1), 130.3 (C2,6), 131.0 (d, J = 9.0 Hz, C2',6'), 134.9 (C1'), 144.9 (C), 161.8 (C4), 165.5 (d, J = 253.5 Hz, C4'), 188.9 (>C=O) ppm. ESI-MS calcd for [M]: 256.09; found: m/z 255.60 [M].
(E)-1-(3'-Fluorophenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (1b): Yield: 41%, white solid, CHFO; R = 0.41 (HEX/EtOAc = 95/5); m.p: 52.6 °C; H-NMR (600 MHz, CDCl): δ = 3.86 (s, 3 H, -OCH), 6.94 (d, J = 8.4 Hz, 2 H, H3,5), 7.28 (m, 1 H, H4'), 7.35 (d, J = 15.6 Hz, 1 H, Hα), 7.47 (m, 1 H, H5′), 7.61 (d, J = 9.0 Hz, 2 H, H2,6), 7.69 (m, 1 H, H2'), 7.79 (d, J = 7.2 Hz, 1 H, H6'), 7.80 (d, J = 15.6 Hz, 1 H, Hβ) ppm C-NMR (150 MHz, CDCl): δ = 55.5 (-OCH), 114.5 (C3,5), 115.2 (d, J = 22.5 Hz, C2'), 119.3 (Cα), 119.5 (d, J = 22.5 Hz, C4'), 124.1 (C6'), 127.5 (C1), 130.2 (d, J = 7.5 Hz, C5'), 130.4 (C2,6), 140.7 (C1'), 145.5 (Cβ), 161.9 (C4), 162.9 (J = 246.0 Hz, C3'), 189.2 (>C=O) ppm. ESI–MS calcd for [M+H]: 257.10; found: m/z 256.90 [M+H].
(E)-1-(3',4'-Difluorophenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (1c): Yield: 43%, white solid, CHFO; R = 0.31 (HEX/EtOAc = 9/1); m.p: 116.7 °C; H-NMR (600 MHz, CDCl): δ = 3.87 (s, 3 H, -OCH), 6.95 (d, J = 9.0 Hz, 2 H, H3,5), 7.29 (m, 1 H, H5′), 7.33 (d, J = 15.6 Hz, 1 H, Hα), 7.61 (d, J = 9.0 Hz, 2 H, H2,6), 7.80 (m, 1 H, H6'), 7.81 (d, J = 15.6 Hz, 1 H, Hβ), 7.86 (m, 1 H, H2') ppm. C-NMR (150 MHz, CDCl): δ = 55.5 (-OCH), 114.6 (C3,5), 117.6 (d, J = 49.5 Hz, C5'), 117.7 (d, J = 49.5 Hz, C2'), 118.6 (Cα), 125.2 (dd, J = 6.0 Hz, J = 3.0 Hz, C6'), 127.3 (C1), 130.4 (C2,6), 135.5 (C1'), 145.7 (Cβ), 162.0 (C4), 187.7 (>C=O) ppm. ESI–MS calcd for [M+H]: 275.09; found: m/z 275.00 [M+H].
(E)-1-(4'-Fluorophenyl)-3-(3-methoxyphenyl)prop-2-en-1-one (2a): Yield 49%, white solid, CHFO [256.09 g/mol]; R = 0.48 (HEX/EtOAc = 9/1); m.p. 66.8 °C; H-NMR (600 MHz, CDCl): δ = 3.86 (s, 3 H, -OCH), 6.98 (dd, J = 7.8 Hz, J = 1.8 Hz, 1 H, H4), 7.15 (dd, J = 2.4 Hz, J = 1.8 Hz, 1 H, H2), 7.18 (dd, J = 9.0 Hz, J = 8.4 Hz, 2 H, H3',5'), 7.23 (d, J = 7.8 Hz, 1 H, H6), 7.34 (dd, J = 8.4 Hz, J = 7.8 Hz, 1 H, H5), 7.47 (d, J = 15.6 Hz, 1 H, Hα), 7.77 (d, J = 15.6 Hz, 1 H, H), 8.05 (dd, J = 9.0 Hz, J = 5.4 Hz, 2 H, H2',6') ppm C-NMR (150 MHz, CDCl): d = 55.4 (-OCH), 113.6 (C2), 115.7 (d, J = 21.0 Hz, C3',5'), 116.4 (C4), 121.1 (C6), 122.0 (Cα), 130.0 (C5), 131.1 (d, J = 9.0 Hz, C2',6'), 134.6 (d, J = 3.0 Hz, C1'), 136.2 (C1), 145.0 (C), 160.0 (C3), 165.7 (d, J = 252.0 Hz, C4'), 188.9 (>C=O) ppm. ESI-MS calcd for [M-HF]: 236.09; found: m/z 235.90 [M-HF].
(E)-1-(3'-Fluorophenyl)-3-(3-methoxyphenyl)prop-2-en-1-one (2b): Yield 42%, yellow solid, CHFO [256.09 g/mol]; R = 0.46 (HEX/EtOAc = 9/1); m.p. 62.4 °C; H-NMR (600 MHz, CDCl): δ = 3.87 (s, 3 H, -OCH), 6.98 (dd, J = 8.4 Hz, J = 2.4 Hz, 1 H, H4), 7.16 (brs, 1 H, H2), 7.25 (d, J = 8.4 Hz, 1 H, H6), 7.29 (m, 1 H, H4'), 7.35 (t, J = 7.8 Hz, 1 H, H5), 7.45 (d, J = 15.6 Hz, 1 H, Hα), 7.50 (m, 1 H, H5'), 7.70 (m, 1 H, H2'), 7.78 (d, J = 9.6 Hz, 1 H, H6'), 7.79 (d, J = 15.6 Hz, 1 H, H) ppm C-NMR (150 MHz, CDCl): d = 55.4 (-OCH), 113.6 (C2), 115.3 (d, J = 22.5 Hz, C2'), 116.6 (C4), 119.8 (d, J = 22.5 Hz, C4'), 121.2 (C6), 121.9 (Cα), 124.2 (C6'), 130.0 (C5), 130.3 (d, J = 7.5 Hz, C5'), 134.5 (C1'), 136.1 (C1), 145.5 (C), 160.1 (C3), 161.1 (d, J = 309 Hz, C3'), 189.6 (>C=O) ppm. ESI-MS calcd for [M+H]: 257.10; found: m/z 257.00 [M+H].
(E)-1-(3',4'-Difluorophenyl)-3-(3-methoxyphenyl)prop-2-en-1-one (2c): Yield 45%, yellow solid, CHFO [274.08 g/mol]; R = 0.44 (HEX/EtOAc = 9/1); m.p. 81.4 °C; H-NMR (600 MHz, CDCl): δ = 3.87 (s, 3 H, -OCH), 6.98 (dd, J = 8.4 Hz, J = 2.1 Hz, 1 H, H4), 7.15 (brs, 1 H, H2), 7.24 (d, J = 7.8 Hz, 1 H, H6), 7.28 (m, 1 H, H5'), 7.35 (t, J = 7.8 Hz, 1 H, H5), 7.42 (d, J = 15.6 Hz, 1 H, Hα), 7.78 (d, J = 15.6 Hz, 1 H, H), 7.81 (m, 1 H, H6'), 7.88 (m, 1 H, H2') ppm C-NMR (150 MHz, CDCl): d = 55.4 (-OCH), 113.6 (C2), 116.6 (C4), 117.6 (d, J = 18.0 Hz, C5'), 117.9 (d, J = 18.0 Hz, C2'), 121.2 (C6), 121.3 (Cα), 125.3 (m, C6'), 130.0 (C5), 135.3 (C1'), 135.9 (C1), 145.7 (C), 160.0 (C3), 187.7 (>C=O) ppm. ESI-MS calcd for [M+H]: 275.09; found: m/z 274.90 [M+H].
(E)-1-(4'-Fluorophenyl)-3-(2,3-dihydroxyphenyl)prop-2-en-1-one (3a): Yield 38%, yellow solid, CHFO [258.07 g/mol]; R = 0.38 (HEX/EtOAc = 7/3); m.p. 132.3 °C; H-NMR (600 MHz, CDOD): δ = 6.75 (t, J = 7.4 Hz, 1 H, H5), 6.87 (dd, J = 7.8 Hz, J = 1.2 Hz, 1 H, H4), 7.19 (dd, J = 7.8 Hz, J = 1.2 Hz, 1 H, H6), 7.28 (dd, J = 9.0 Hz, J = 8.4 Hz, 2 H, H3',5'), 7.79 (d, J = 15.6 Hz, 1 H, Hα), 8.14 (dd, J = 9.0 Hz, J = 5.4 Hz, 2 H, H2',6'), 8.15 (d, J = 15.6 Hz, 1 H, H) ppm C-NMR (150 MHz, CDOD): d = 116.6 (d, J = 21.0 Hz, C3',5'), 118.0 (C4), 120.6 (C5), 121.0 (C6), 122.4 (Cα), 123.2 (C1), 132.4 (d, J = 9.0 Hz, C2',6'), 136.2 (C1'), 142.8 (C), 146.8 (C3), 147.7 (C2), 166.1 (d, J = 264.0 Hz, C4'), 191.6 (>C=O) ppm. ESI-MS calcd for [M+H]: 259.08; found: m/z 258.90 [M+H].
(E)-1-(3'-Fluorophenyl)-3-(2,3-dihydroxyphenyl)prop-2-en-1-one (3b): Yield 35%, yellow solid, CHFO [258.07 g/mol]; R = 0.28 (HEX/EtOAc = 8/2); m.p. 122.8 °C; H-NMR (600 MHz, CDOD): δ = 6.75 (dd, J = 8.4 Hz, J = 7.8 Hz, 1 H, H5), 6.87 (dd, J = 7.8 Hz, J = 1.2 Hz, 1 H, H4), 7.20 (dd, J = 8.4 Hz, J = 1.2 Hz, 1 H, H6), 7.39 (m, 1 H, H4'), 7.59 (m, 1 H, H5'), 7.75 (m, 1 H, H2'), 7.78 (d, J = 15.6 Hz, 1 H, Hα), 7.89 (m, 1 H, H6'), 8.15 (d, J = 15.6 Hz, 1 H, H) ppm C-NMR (150 MHz, CDOD): d = 115.9 (d, J = 22.5 Hz, C2'), 118.1 (C4), 120.6 (C5), 120.7 (d, J = 18.0 Hz, C4'), 121.1 (C6), 122.4 (Cα), 123.1 (C1), 125.5 (d, J = 3.0 Hz, C6'), 131.7 (d, J = 9.0 Hz, C5'), 142.1 (d, J = 6.0 Hz, C1'), 143.4 (C), 146.8 (C3), 147.8 (C2), 163.6 (d, J = 301.5.0 Hz, C3'), 191.7 (>C=O) ppm. ESI-MS calcd for [M+H]: 259.08; found: m/z 258.80 [M+H].
(E)-1-(3',4'-Difluorophenyl)-3-(2,3-dihydroxyphenyl)prop-2-en-1-one (3c): Yield 44%, yellow solid, CHFO [276.06 g/mol]; R = 0.22 (HEX/EtOAc = 8/2); m.p. 144.2 °C; H-NMR (600 MHz, CDOD): δ = 6.75 (dd, J = 8.4 Hz, J = 7.8 Hz, 1 H, H5), 6.88 (dd, J = 7.8 Hz, J = 1.8 Hz, 1 H, H4), 7.20 (dd, J = 8.4 Hz, J = 1.2 Hz, 1 H, H6), 7.46 (m, 1 H, H5'), 7.79 (d, J = 15.6 Hz, 1 H, Hα), 7.95 (m, 1 H, H6'), 7.97 (m, 1 H, H2'), 8.17 (d, J = 15.6 Hz, 1 H, H) ppm C-NMR (150 MHz, CDOD): d = 118.1 (C4), 118.6 (d, J = 22.5 Hz, C5'), 118.7 (d, J = 21.0 Hz, C2'), 120.6 (C5), 121.1 (C6), 121.8 (Cα), 123.1 (C1), 126.9 (d, J = 9.0 Hz, C6'), 137.2 (C1'), 143.4 (C), 146.8 (C3), 147.8 (C2), 150.8 (C3',4'), and 190.2 (>C=O) ppm. ESI-MS calcd for [M+H]: 277.07; found: m/z 276.80 [M+H].
(E)-1-(4'-Fluorophenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (4a): Yield 38%, yellow solid, CHFO [242.07 g/mol]; R = 0.51 (HEX/EtOAc = 9/1); m.p. 187.4 °C; H-NMR (600 MHz, CDCl): δ = 5.64 (1 H, -OH), 6.89 (d, J = 8.4 Hz, 2 H, H3,5), 7.17 (dd, J = 9.0 Hz, J = 8.4 Hz, 2 H, H3',5'), 7.38 (d, J = 15.6 Hz, 1 H, Hα), 7.56 (d, J = 8.4 Hz, 2 H, H2,6), 7.78 (d, J = 15.6 Hz, 1 H, H), 8.05 (dd, J = 9.0 Hz, J = 5.4 Hz, 2 H, H2',6') ppm C-NMR (150 MHz, CDCl): d = 115.7 (d, J = 27.0 Hz, C3',5'), 116.1 (C3,5), 119.3 (Cα), 127.6 (C1), 130.5 (C2,6), 131.0 (d, J = 10.5 Hz, C2',6'), 134.8 (C1'), 145.1 (C), 158.2 (C4), 189.2 (>C=O) ppm. ESI-MS calcd for [M-H]: 241.06; found: m/z 240.80 [M-H].
(E)-1-(3'-Fluorophenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (4b): Yield 44%, yellow solid, CHFO [242.07 g/mol]; R = 0.60 (HEX/EtOAc = 8/2); m.p. 63.7 °C; H-NMR (600 MHz, CDCl): δ = 6.90 (d, J = 9.0 Hz, 2 H, H3,5), 7.29 (m, 1 H, H4'), 7.34 (d, J = 15.6 Hz, 1 H, Hα), 7.48 (m, 1 H, H5'), 7.56 (d, J = 8.4 Hz, 2 H, H2,6), 7.68 (d, J = 9.6 Hz, 1 H, H2'), 7.79 (d, J = 15.0 Hz, 1 H, H), 7.80 (d, J = 6.0 Hz, 1 H, H6') ppm C-NMR (150 MHz, CDCl): d = 115.2 (d, J = 21.0 Hz, C2'), 116.1 (C3,5), 119.3 (Cα), 119.6 (d, J = 21.0 Hz, C4'), 124.1 (d, J = 3.0 Hz, C6'), 127.5 (C1), 130.2 (d, J = 7.5 Hz, C5'), 130.7 (C2,6), 140.6 (d, J = 6.0 Hz, C1'), 145.6 (C), 158.4 (C4), 162.5 (d, J = 246.0 Hz, C3'), 189.5 (>C=O) ppm. ESI-MS calcd for [M-H]: 241.06; found: m/z 240.80 [M-H].
(E)-1-(3',4'-Difluorophenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (4c): Yield 33%, yellow solid, CHFO [260.06 g/mol]; R = 0.48 (HEX/EtOAc = 7/3); m.p. 176.8 °C; H-NMR (600 MHz, CDCl): δ = 5.60 (1 H, -OH), 6.90 (d, J = 8.4 Hz, 2 H, H3,5), 7.29 (m, 1 H, H5'), 7.32 (d, J = 15.6 Hz, 1 H, Hα), 7.57 (d, J = 8.4 Hz, 2 H, H2,6), 7.79 (m, 1 H, H6'), 7.80 (d, J = 15.6 Hz, 1 H, H), 7.86 (m, 1 H, H2') ppm C-NMR (150 MHz, CDCl): d = 116.1 (C3,5), 117.4 (d, J = 21.0 Hz, C5'), 117.7 (d, J = 22.5 Hz, C2'), 118.6 (Cα), 125.2 (d, J = 9.0 Hz, C6'), 127.5 (C1), 130.7 (C2,6), 135.5 (C1'), 145.7 (C), 158.4 (C4), and 187.9 (>C=O) ppm. ESI-MS calcd for [M+H]: 261.07; found: m/z 260.90 [M+H].
In vitro cytotoxicity of fluorinated chalcones against HepG2 cells
The in vitro cytotoxic activity of the fluorinated chalcones was assessed against HepG2 cancer cells by determining their half-maximal inhibitory concentration (IC, µM). Doxorubicin was used as a positive control for the MTT assay. To determine the IC values, the percent inhibition of chalconoids was evaluated at final concentrations of 2, 8, 32, and 128 µg/mL in the wells. As summarized in
Isolated yields of fluorinated chalconoids synthesized by grinding and microwave-assisted procedures and their cytotoxic activity against HepG2 cells
|
Fluorinated chalcone |
Yield (%) |
IC50±SD (µM) | |
|
|
Grinding |
Microwave |
|
|
1a |
42 |
(*) | |
|
1b |
41 |
(*) | |
|
1c |
43 |
(*) | |
|
2a |
49 |
67.51±2.26 | |
|
2b |
42 |
72.51±1.79 | |
|
2c |
45 |
68.37±8.75 | |
|
3a |
38 |
87.22±8.52 | |
|
3b |
35 |
90.55±2.94 | |
|
3c |
44 |
79.22±5.03 | |
|
4a |
38 |
105.59±4.05 | |
|
4b |
44 |
92.86±3.88 | |
|
4c |
33 |
108.20±7.92 | |
|
Doxorubicin as a positive control |
28.70±2.21 | ||
DISCUSSION
Chemistry
Fluorinated chalcones were successfully synthesized via a cross-aldol condensation reaction between benzaldehydes and fluorinated acetophenones in the presence of sodium hydroxide or montmorillonite K10 as catalysts. Starting with the grinding method under solvent-free conditions, an equimolecular mixture of starting materials was ground for 10 min at room temperature before solid NaOH was added. The mixture was further ground for 20 min to afford the expected chalcones 1a-1c and 2a-2c in moderate yields of 41-49%. As reported in our previous results, the isolated yields of chalcones synthesized by a base-catalyzed Claisen-Schmidt condensation reaction of aromatic benzaldehydes with acetophenones were dependent on the substituents in the aromatic rings8. Under basic conditions, the C‒C bond was formed by nucleophilic attack from the enolate anion to the aldehyde group. Therefore, the presence of electron-donating groups on the benzaldehyde ring was unfavorable for the C‒C coupling reaction. Herein, the moderate conversions of chalcones can be attributed to the effects of the substituents on the aromatic rings. In addition, the yields of the desired chalcones were found to be affected by the synthetic procedure, which led to low yields of the desired chalcones. We found that the majority of the Michael addition product formed by the condensation of the expected chalcone with a second enolate form of acetophenone was observed when the reagents and solid NaOH were ground together. The Michael products were isolated as the main products, and their NMR data are not shown in the present results.
Our target focused on the synthesis of chalcones bearing –F groups in ring A, while the –OCH and –OH groups were substituted in the B ring moiety. In accordance with our previous results of the synthesis of hydroxy chalcones by the grinding method, the C‒C coupling reaction under NaOH-catalyzed conditions was inefficient because of the effects of hydroxy groups8. This observation was attributed to the formation of a phenoxide anion, which significantly decreased the reactivity of the carbonyl group via resonance effects. Indeed, our efforts failed to prepare hydroxy chalcones under basic conditions. Clearly, the need for the synthesis of hydroxy chalcones without the protection of hydroxy functional groups becomes apparent. The available protocol for the preparation of the required chalcones under microwave irradiation uses montmorillonite K10 clay as a solid catalyst7. Regardless of the absence of hydroxy chalcones in a series of synthesized chalcones, we decided to apply this procedure to synthesize hydroxy chalcones. Surprisingly, except for 4-hydroxy/2,3-dihydroxybenzaldehydes, when a mixture including 2-hydroxy/3-hydroxy/3,4-dihydroxybenzaldehydes and fluorinated acetophenones as starting materials was irradiated at 150 °C for 1.5 h, the expected chalcones were not isolated regardless of the coupling reactions being repeated. We also tested different synthetic conditions for Claisen-Schmidt condensation (irradiation time: 2.0 h; temperature: 170 C) but obtained disappointing results after purification.
Clay-based catalysts for C‒C bond formation have been widely utilized in organic synthesis21, 22, 23. The metal ions (M) present in the MK10 catalyst function as Lewis acids, increasing the electrophilicity of the carbonyl group via coordination7. As a result, the attack of the aldehyde group by the enol form of acetophenone becomes favorable for the formation of the desired chalcones. After our attempts, fluorinated chalcones (3a-3c, 4a-4c) were obtained in yields not exceeding 44%.
The chemical structures of the fluorinated chalcones were identified via H NMR, C NMR and MS analyses. The H-NMR spectra revealed doublet signals between 7.3 and 7.8 ppm with coupling constants of approximately 15 Hz, indicating the trans-configuration of the chalcones.
Cytotoxicity to HepG2 cells
To evaluate the anticancer activities of the fluorinated chalconoids, we tested the effects of the synthesized compounds on the growth of HepG2 cancer cells. Concentration‒response experiments were carried out to obtain IC (µM) values. As shown in
In general, the biological responses of chalcones are attributed to the presence of the α, -unsaturated carbonyl scaffold as a Michael acceptor. Biologically, the formation of covalent bonds between the α, -unsaturated carbonyl group and nucleophiles such as the sulfhydryl of cysteine or other thiols (-SH) in biological systems to obtain the Michael adduct plays a crucial role in the inhibitory activities31, 32, 33, 34, 35. Indeed, structure‒activity relationship (SAR) analysis of a series of chalcone-based compounds indicated that saturation of the alkene moiety in the chalcone skeleton completely abolished the anticancer activity33. In other words, the cytotoxic activities of chalcones toward cancer cells may be due to the Michael acceptor mechanism (Figure 3).

Michael addition of a chalcone with thiol nucleophiles in biological systems. The picture was taken from Ref. 35.
A comparison of the chemical structures of all the fluorinated chalcones revealed that the presence of fluoro groups in the A ring and the 4-OCH substitution in the B ring quenched the anticancer activities of compounds 1a-1c, whereas the remaining candidates presented antiproliferative activities. The effects of fluoro-containing molecules on biological activities remain a trial-and-error process. SAR analysis is relatively simple for interpreting the influence of the fluorine substituent14, 15, 36. In this study, the motif of chalcones 1a-1c abolished the cytotoxic effects on HepG2 cells. In our synthetic strategy, the A ring of chalcones is substituted with fluoro groups, while methoxy/hydroxy groups are introduced to the B ring. Considering the contribution of fluoro groups in the A ring to the anticancer activity, compound 2a (IC = 67.51±2.26 µM) displayed a slight increase in activity compared with that of 2b (IC = 72.51±1.79 µM), suggesting that the 4'-F scaffold might contribute to the inhibition of HepG2. The IC values of compounds 3a (IC = 87.22±8.52 µM) and 3c (IC = 79.22±5.03 µM) also suggested the effect of the 4'-F motif on anticancer activity. In our current work, fluorinated chalcones (2a-2c) bearing a 3-OCH group in the B ring demonstrated higher activities than 3a-3c did. The 3-hydroxy group and/or increased hydroxylation of the B ring had no apparent effect on the activity against HepG2 cells. 4-Hydroxy chalcone analogs (4a-4c) exhibited lower activities, suggesting the importance of the 3-OCH/OH position in the inhibitory activity against HepG2 cells.
CONCLUSIONS
In summary, twelve fluoro-containing chalcones were synthesized in 33--49% yields via Claisen-Schmidt condensation by grinding and microwave-assisted methods in a solvent-free environment. Among the structures synthesized via eco-friendly approaches, four fluorinated chalcones, 3a-3c and 4c, are novel compounds. The chemical structures of chalcones were elucidated via NMR and MS analyses. All the compounds were assessed for their anticancer activity against HepG2 cancer cells. Most fluorinated chalcones, with IC values in the range of 67.51--108.20 µM, showed anticancer activity, except for 1a-1c. The scaffold of 4'-F-3-OCH might contribute to the cytotoxicity against HepG2 cells and warrants further investigation.
LIST OF ABBREVIATIONS
brs: Broad singlet
CC: Column chromatography
DCM: dichloromethane
d: Doublet
dd: Doublet of doublets
DMSO-d: Dimethyl sulfoxide-d
equiv: Equivalent
EtOAc: Ethyl acetate
ESI: Electrospray ionization
HEX:n-Hexane
HepG2: Human hepatocarcinoma cell line
IC: Half-maximal inhibitory concentration
MK10: Montmorillonite K10
NMR: Nuclear magnetic resonance
MS: Mass spectrometry
m: Multiplet
RT: Room temperature
SAR: Structure-Activity Relationship
s: Singlet
t: Triplet
TLC: Thin layer chromatography
td: triplet of doublets,
UV: Ultraviolet
AUTHOR CONTRIBUTIONS
Conceptualization: Hoang Minh Hao; development of the synthetic methods: Ho Phuong and Hoang Minh Hao; synthesis of fluorinated chalcones: Ho Phuong, Nguyen Thi Ngoc Anh and Hoang Minh Hao; structural assignment via NMR, MS spectra: Ho Phuong and Hoang Minh Hao; HepG2 assay: Than Quoc An Ha; analysis of cytotoxicity basing on structure: Hoang Minh Hao, Nguyen Thi Loan and Than Quoc An Ha; writing original draft preparation: Ho Phuong and Hoang Minh Hao; writing-review and editing: Hoang Minh Hao. All authors have read and agreed to the published version of the manuscript.
COMPETING INTERESTS
The authors declare that they have no competing interests.
SUPPORTING INFORMATION
The NMR and MS spectra of all the fluorinated chalcones can be found in the Supporting Information.
ACKNOWLEDGMENT
This work belongs to the project grant No: T2024-168 funded by Ho Chi Minh City University of Technology and Education, Vietnam.