

Synthesis and in vitro α-glucosidase inhibitory activity of some of the derivatives of 3-methylrhodanine
- Faculty of Chemistry, University of Science, Vietnam National University – Ho Chi Minh City
Abstract
In the further research and synthesis of highly bioactive 3-methylrhodanine derivatives, the preparation of benzylidene rhodanines followed by bicyclization to form pyrazole compounds has been undertaken. Using a convenient and simple method, twelve compounds have been synthesized including six benzylidene rhodanine derivatives ranging from a good to excellent yield, with four phenylhydrazone and two pyrazole derivatives at a low yield. The structures of all synthesized compounds were elucidated by spectral methods of analysis. Among them, six heterocyclic compounds, namely 4-(2-phenylhydrazono)-5-benzylidene-3-methylthiazolidine-2-thione (3'a-3'd), 3-(4-nitrophenyl)-2-phenyl-2,6-dihydro-5H-pyrazolo[3,4-d]thiazole-5-thione (3''e), and 3-(4-cyanophenyl)-2-phenyl-2,6-dihydro-5H-pyrazolo[3,4-d]thiazole-5-thione (3''f) were synthesized for the first time. The synthesized compounds were evaluated in vitro against α-glucosidase and exhibited promising antidiabetic activity with IC50 values ranging from 11.3 to 21.9 µM.
INTRODUCTION
Rhodanine (2-thioxo-1,3-thiazolidin-4-one) (Figure 1) is a heterocyclic compound belonging to the thiazolidinone family. In recent years, rhodanine and its derivatives have been widely studied by many research groups due to their numerous bioactivities such as anticancer, antibacterial, antifungal, anticonvulsant, anti-inflammatory, antiviral, and antioxidant 1, 2. Besides this, many studies on structure-activity relationships have also been performed to conduct their analogues regarding the drug development process2, 3. Additionally, rhodanine derivatives could be more interesting due to the fact that they might be starting material for the preparation of other biologically active compounds. Some synthetic rhodanines show potential as drugs for use in medical treatments. Among them, 5-benzylidene-2-thioxothiazolidin-4-one derivatives have been widely studied because of their rich biological properties and broad applicability in medicine. In 2018, 4-[2-(2-chlorophenoxy)ethoxy]benzylidene-2-thioxothiazolidin-4-one (Figure 1) showed a high capacity in the treatment of diabetes mellitus (roughly equivalent to the standard Osiglitazone drug)4. Epalrestat or 2-[(2-methyl-3-phenylallylidene)-4-oxo-2-thioxothiazolidin-3-yl]acetic acid (Figure 1)in particular, which was synthesized in the same year, has been shown to have antidiabetic activity and has been approved for marketing as a therapeutic drug for the treatment of diabetic complications5.

Structure of rhodanine and rhodanine derivatives with good antidiabetic activity
3-Methylrhodanine derivatives have long been studied and are known to possess various biological activities such as antidiabetic6 and antimicrobial7, 8. Some studies have also shown that rhodanine can be used as an adsorbing agent, allowing for the separation of Au, Ag, and Pt(9). Furthermore, pyrazoline is proven to have anti-cancer9 and anti-inflammatory activity10. However, there are currently no studies on the synthesis of rhodamine derivatives in the form of an adjacent double ring. This study will focus on the synthesis of heterocyclic compounds from 3-methylrhodanine in the form of phenylhyrazone, pyrazole, and pyrazoline and the testing of the antidiabetic activity of the synthesized compound toward the enzyme -glucosidase.
MATERIALS AND METHODS
General
The 3-methylrhodanine was prepared by Prof. Fritz Duus (University of Roskilde, Denmark). Benzaldehyde (assay 98%) was purchased from Sigma-Aldrich. 4-methylbenzaldehyde (assay 97%), 4-methoxybenzaldehyde (assay 97%), 4-chlorobenzaldehyde (assay 97%), 4-nitrobenzaldehyde (assay 97%), 4-nitrobenzaldehyde (assay 97%), 4-cyanobenzaldehyde (assay 97%) and phenylhydrazine (assay 97%) were obtained from Acros Organics. Sodium acetate, ethyl acetate (purity 99.5%), -hexane (purity 99.5%), methanol and chloroform (purity 99%), and glacial acetic acid were obtained from Xilong Chemical Co., Ltd (China). -Glucosidase (EC 3.2.1.20) from (750 UN) and -nitrophenyl--ᴅ-glucopyranoside, NaHPO, NaHPO and NaCO were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Acarbose and dimethylsulfoxide were purchased from Merck (Darmstadt, Germany). The chemicals and organic solvents were obtained from commercial sources and used without further purification.
The NMR spectra were acquired using the Bruker Avance III spectrometer (500 MHz for H and 125 MHz for C). Chemical shifts are expressed in parts per million (ppm) and reported relative to the residual solvent signal as an internal reference. High resolution mass spectra (HRMS) were recorded using a HRMS X500 QTOF mass spectrometer in electrospray ionization (ESI) mode. The absorbance (OD) was measured using a Shimadzu UV-1800 UV-VIS spectrophotometer (Shimadzu PTE. LTD., Singapore). The melting points were determined using a Gallenkamp digital Melting point apparatus 5A-6797 with a rate of heating of 2°C/min. Reactions were monitored using thin layer chromatography (TLC) on silica gel plates (silica gel 60 F, Merck), visualized under ultraviolet light (254 nm). Column chromatography was performed on silica gel Merck 60 (230–400 mesh) purchased from HiMedia Laboratories Pvt. Ltd. (India).
General procedure for the synthesis of 5-arylidene-3-methylrhodanine compounds 2a–2f
To a stirred solution of 3-methylrhodanine 1 (735 mg, 5 mmol, 1eq) and sodium of acetate (820 mg, 10 mmol, 2eq) in MeOH: AcOH (5:2) (15 mL) at 80 °C, substituted aldehyde (20 mmol, 4eq) was added. The reaction was monitored using TLC. After 4 hours of stirring, the precipitate obtained was filtered and recrystallized in ethyl acetate to give the corresponding products 2a–2f.
General procedure for the synthesis of compounds 3'a–3'd and 3''e–3''f
A mixture of 5-(4-alkylbenzylidene)-3-methyl-2-thioxothiazolidin-4-one) 2a–2f (1 mmol, 1eq), sodium acetate (246 mg, 3 mmol, 3eq), and phenylhydrazine (196 µL, 2 mmol, 2eq) in ethanol (10 mL) was placed in a 50 mL round-bottomed flask. The mixture was stirred for 30 minutes at room temperature and then slowly heated to 80 °C. The reaction was monitored using TLC. After 4 hours of stirring at 80 °C, a solution HCl 10% (20 mL) was added to the mixture. The two distinct phases were separated. The aqueous phase was extracted with ethyl acetate (3 x 20mL). All organic phases were collected, dried over NaSO and concentrated under reduced pressure. The obtained crude product was purified by column chromatography using eluent hexane:chloroform (95:5) to give the corresponding products 3'a–3'd and 3''e–3''f.
Biological tests
The enzyme -glucosidase inhibitory activity is based on photometric analysis. The enzyme -glucosidase catalyzes the hydrolysis of heterogenous substrates such as sucrose and -nitrophenyl--ᴅ-glucopyranoside. Therefore, to investigate the inhibitory activity of enzyme -glucosidase, -nitrophenyl--ᴅ-glucopyranoside (-NP-G) was used as the starting substrate and the enzyme -glucosidase converts this substrate into -ᴅ-glucose and -nitrophenol (Figure 2). During the reaction, the amount of -ᴅ-glucose produced and the amount of -nitrophenol formed are proportional to the hydrolyzed -NP-G. Therefore, the amount of -ᴅ-glucose produced as well as the amount of -nitrophenol formed can be quantified based on the absorbance of -nitrophenolate at 401 nm.

The hydrolysis of
When the sample contains the -glucosidase inhibitor, the amount of -nitrophenol formed will decrease. Comparing the color intensity of the solution with and without an inhibitor will enable the calculation of the ability to inhibit the enzyme -glucosidase of that sample. The ability to inhibit the enzyme -glucosidase was evaluated by the percentage inhibition value (I %):
In which:
A: Value of the optical density of the solution that does not contain the survey sample.
A: Value of the optical density of the solution containing the survey sample.
Based on the percentage inhibition at different concentrations of the sample, the ability of the sample to inhibit the enzyme -glucosidase was evaluated through its IC value. The IC value was defined as the concentration of a sample that inhibited 50% of the -glucosidase activity. Lastly, acarbose, a well-known α-glucosidase inhibitor, was successfully used as a positive control.
The antidiabetic activity of the synthesized compounds was evaluated in the Laboratory of Pharmaceutical Chemistry, Faculty of Biology, University of Science, Vietnam National University – Ho Chi Minh City.
The inhibitory activity of -glucosidase was determined according to the modified method of Kim et al. 11. The test samples were dissolved in DMSO at a concentration of 0.1 g/mL and diluted with phosphate buffer to the test concentrations (μM). 3 mM -nitrophenyl--ᴅ-glucopyranoside (25 μL) and 0.2 U/mL -glucosidase (25 μL) in 0.01 M phosphate buffer (pH = 7.0) were added to the sample solution (625 μL) to start the reaction. Each reaction was carried out at 37 °C for 30 min and stopped by adding 0.1 M NaCO (375 μL). The enzymatic activity was quantified by measuring absorbance at a 401 nm wavelength. The amount of enzyme liberating -nitrophenol (1.0 μM) per min corresponds to one unit of -glucosidase activity.
Each test sample was performed at five concentrations of 250, 100, 50, 25, and 10 µM, three times each. For each concentration of sample, a blank sample was also performed. The blank sample was the same as the test sample but the α-glucosidase enzyme solution was replaced with a 0.01 M phosphate buffer with a pH of 7.0. The percentage inhibition value (I %) of each concentration tested was the average value of three values of optical density measured at each concentration.
All experiments were performed completely independently and repeated three times. Statistical analysis was performed using Microsoft Excel 365 software. Using one-way analysis of variance (ANOVA), the results were calculated based on the mean with a standard deviation showing statistical significance (p < 0.05).
RESULTS
The 5-arylidene-3-methylrhodanine derivatives were synthesized using the general procedure above. We obtained six derivatives with yields ranging from good to excellent (Figure 3). The chemical structures of these products were elucidated using the H and C NMR spectra.
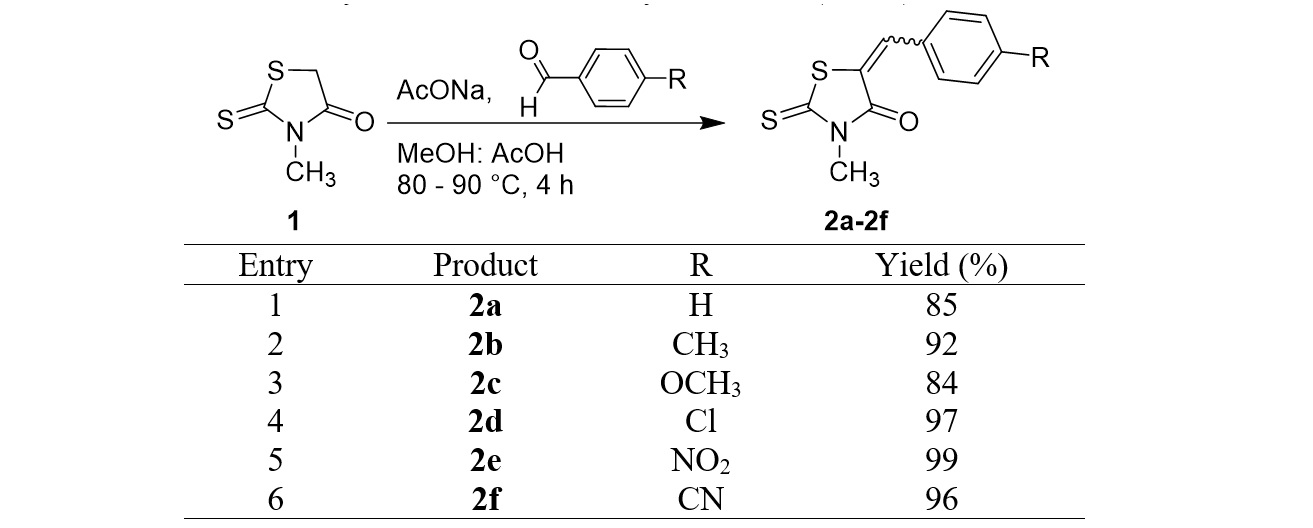
Synthesis of the 3-methylrhodanine (2a–2f) derivatives
5-Benzylidene-3-methyl-2-thioxothiazolidin-4-one (2a): 1.00 g, yield: 85%, yellow crystals, m.p. 168–170 °C. H-NMR (500 MHz, CDCl) (ppm): 7.75 (, 1H), 7.43 – 7.54 (, 5H), 3.53 (, 3H); C-NMR (125 MHz, CDCl) (ppm): 193.6, 167.8, 133.4, 133.2, 130.8, 130.6, 129.4, 123.3, 31.2.
5-(4-Methylbenzylidene)-3-methyl-2-thioxothiazolidin-4-one (2b): 1.15 g, yield: 92%, yellow crystals, m.p.170–171 °C. H-NMR (500 MHz, CDCl) (ppm): 7.73 (, 1H), 7.40 (, = 8.1 Hz, 2H), 7.29 (, = 8.0 Hz, 2H), 3.52 (, 3H), 2.41 (, 3H); C-NMR (125 MHz, CDCl) (ppm): 193.8, 168.0, 141.8, 137.8, 133.5, 130.9, 130.3, 122.1, 31.3, 21.8.
5-(4-Methoxybenzylidene)-3-methyl-2-thioxothiazolidin-4-one (2c): 1.11 g, yield: 84%, yellow crystals, m.p. 181–183 °C. H-NMR (500 MHz, CDCl) (ppm): 7.71 (, 1H), 7.47 (, = 8.3 Hz, 2H), 7.00 (, = 8.3 Hz, 2H), 3.88 (, 3H), 3.52 (, 3H); C-NMR (125 MHz, CDCl) (ppm): 193.6, 168.0, 161.9, 133.3, 132.8, 126.1, 120.3, 115.0, 55.6, 31.3.
5-(4-Chlorobenzylidene)-3-methyl-2-thioxothiazolidin-4-one (2d): 1.31 g, yield: 97%, yellow crystals, m.p. 202–203 °C. H-NMR (500 MHz, CDCl) (ppm): 7.68 (, 1H), 7.46 (, J = 8.7 Hz, 2H), 7.43 (, J = 8.7 Hz, 2H), 3.53 (, 3H); C-NMR (125 MHz, CDCl) (ppm): 193.1, 167.8, 137.1, 132.0, 131.8, 131.6, 129.8, 124.0, 31.4.
5-(4-Nitrobenzylidene)-3-methyl-2-thioxothiazolidin-4-one (2e): 1.39 g, yield: 99%, yellow crystals, m.p. 156–157 °C. H-NMR (500 MHz, CDCl) (ppm): 8.33 (, = 8.8 Hz, 2H), 7.75 (, 1H), 7.65 (, = 8.7 Hz, 2H), 3.55 (, 3H); C-NMR (125 MHz, CDCl) (ppm): 192.2, 167.5, 148.2, 139.4, 131.1, 129.5, 128.0, 124.6, 31.6.
5-(4-Cyanobenzylidene)-3-methyl-2-thioxothiazolidin-4-one (2f): 1.25 g, yield: 96%, yellow crystals, m.p. 145–147 °C. H-NMR (500 MHz, CDCl) (ppm): 7.76 (, = 8.1 Hz, 2H), 7.70 (, 1H), 7.59 (, = 8.1 Hz, 2H), 3.54 (, 3H); C-NMR (125 MHz, CDCl) (ppm): 192.3, 167.6, 137.6, 133.1, 130.8, 130.0, 127.3, 118.1, 113.8, 31.5.

The synthetic route for the preparation of 3-methylrhodanine derivatives
The cyclization of 5-arylidene-3-methylrhodanine compounds 2a–2f with phenylhydrazine was archived with different results depending on the substituents (Figure 4). We obtained the phenylhydrazone 3'a–3'd and pyrazole 3''e–3''f derivatives instead of the desired pyrazoline derivatives 3a–3f (Figure 4). The chemical structures of these obtained products were elucidated by H and C NMR as well as HR-MS spectra.
4-(2-Phenylhydrazono)-5-benzylidene-3-methylthiazolidine-2-thione (3'a): 45 mg, yield: 14%, orange crystals, m.p. 154–155 °C. 1H-NMR (500 MHz, CDCl3) δH (ppm): 7.76 (, 1H), 7.57 (, J = 7.5 Hz, 2H), 7.47 (, J = 7.8, 7.3 Hz, 2H), 7.40 (, J = 7.4, 1.1 Hz, 1H), 7.28 (, J = 8.4, 7.4 Hz, 2H), 7.01 (, J = 8.6, 1.2 Hz, 2H) , 6.93 (, J = 7.4, 1.1 Hz, 1H), 6.29 (, 1H), 3.46 (, 3H); 13C-NMR (125 MHz, CDCl3) δC (ppm): 190.2, 159.9, 146.6, 134.0, 130.8, 130.5, 130.1, 129.9, 129.3, 129.2, 121.2, 114.1, 29.8. HRMS (ESI): m/z calcd for CHNS [M-CH]: 310.0473; found: 310.0487.
4-(2-Phenylhydrazono)-5-(4-methylbenzylidene)-3-methylthiazolidine-2-thione (3'b): 34 mg, yield: 10%, orange crystals, m.p. 155–158 °C. H-NMR (500 MHz, CDCl) (ppm): 7.73 (, 1H), 7.46 (, = 8.1 Hz, 2H), 7.28 (, = 8.4, 7.6 Hz, 4H), 7.01 (, = 8.3, 0.9 Hz, 2H), 6.92 (, J = 7.3, 1.1 Hz, 1H), 6.28 (, 1H), 3.44 (, 3H), 2.40 (, 3H); C-NMR (125 MHz, CDCl) (ppm): 189.5, 166.2, 146.6, 140.5, 131.2, 130.6, 130.1, 130.0, 129.3, 121.1, 120.1, 114.1, 29.7, 21.7. HRMS (ESI): m/z calcd for CHNS [M-CH]: 324.0629; found: 324.0647.
4-(2-Phenylhydrazono)-5-(4-methoxybenzylidene)-3-methylthiazolidine-2-thione (3'c): 25 mg, yield: 7%, orange crystals, m.p. 161–162 °C. H-NMR (500 MHz, CDCl) (ppm): 7.72 (, 1H), 7.52 (, = 8.8 Hz, 2H), 7.28 (, = 8.4, 7.4 Hz, 2H), 7.01 (, = 8.6, 1.0 Hz, 2H), 6.99 (, = 8.7 Hz, 2H), 6.92 (, J = 7.4, 1.1 Hz, 1H), 6.27 (, 1H), 3.86 (, 3H), 3.44 (, 3H); C-NMR (125 MHz, CDCl) (ppm): 194.6, 166.3, 161.0, 147.5, 146.7, 131.9, 130.4, 129.3, 126.7, 121.1, 114.8, 114.1, 55.6, 29.7. HRMS (ESI): m/z calcd for CHNOS [M-CH]: 340.0578; found: 340.0595.
4-(2-Phenylhydrazono)-5-(4-chlorobenzylidene)-3-methylthiazolidine-2-thione (3'd): 30 mg, yield: 8%, orange crystals, m.p. 142–143 °C.H-NMR (500 MHz, CDCl) (ppm): 7.69 (, 1H), 7.49 (, = 8.7 Hz, 2H), 7.43 (, = 8.7 Hz, 2H), 7.28 (, = 8.6, 7.3 Hz, 2H), 7.01 (, = 8.6, 1.0 Hz, 2H), 6.93 (, J = 7.3, 1.1 Hz, 1H), 6.31 (, 1H), 3.45 (, 3H);C-NMR (125 MHz, CDCl) (ppm): 197.4, 165.8, 146.5, 135.9, 132.5, 131.2, 129.5, 129.4, 129.1, 121.9, 121.3, 114.3, 29.9. HRMS (ESI): m/z calcd for CHClNS [M-CH]: 344.0083; found: 344.0109 and CHClNS [M-CH]: 346.0053; found: 346.0091.
3-(4-Nitrophenyl)-2-phenyl-2.6-dihydro-5H-pyrazolo[3.4-d]thiazole-5-thione (3''e): 25 mg, yield: 7%, orange crystals, m.p. 156–157 °C.H-NMR (500 MHz, CDCl) (ppm): 8.22 (, = 8.8 Hz, 2H), 7.78 (, = 8.9 Hz, 2H), 7.70 (, 1H), 7.32 (, = 8.6, 7.4 Hz, 2H), 7.15 (, = 8.7, 1.0 Hz, 2H), 6.95 (, = 7.3, 0.9 Hz, 1H);C-NMR (125 MHz, CDCl) (ppm): 188.4, 147.3, 143.8, 141.9, 134.0, 129.6, 126.7, 126.4, 124.3, 121.5, 114.4, 113.3. HRMS (ESI): m/z calcd for CHKNOS [M+K]: 392.9882; found: 392.9923.
3-(4-Cyanophenyl)-2-phenyl-2.6-dihydro-5H-pyrazolo[3.4-d]thiazole-5-thione (3''f): 18 mg, yield: 5%, orange crystals, m.p. 145–147 °C.H-NMR (500 MHz, CDCl) (ppm): 7.72 (, = 8.4 Hz, 2H), 7.66 (, 1H), 7.63 (, = 8.4 Hz, 2H), 7.31 ( , = 8.5, 7.4 Hz, 2H), 7.13 (, = 8.6, 0.9 Hz, 2H), 6.93 (, = 7.3, 1.0 Hz, 1H);C-NMR (125 MHz, CDCl) (ppm): 211.1, 169.0, 143.9, 139.9, 134.5, 132.5, 129.6, 126.4, 121.2, 119.2, 114.3, 113.2, 111.2. HRMS (ESI): m/z calcd for CHNOS [M+HO]: 352.0453; found: 352.0477.
The synthesized compounds including benzylidene rhodanines 2a–2f, phenylhydrazones 3'a–3'd and pyrazoles 3''e–3''f were evaluated for their antidiabetic activity against the enzyme -glucosidase with Acarbose as the positive control. The inhibition (I %) and IC values are shown in
|
Entry |
Compound |
Inhibition (I %) |
IC50 (µM) | ||||
|
250 (µM) |
100 (µM) |
50 (µM) |
25 (µM) |
10 (µM) | |||
|
1 |
1 |
* |
53,4 ± 1,5 |
25,5 ± 1,7 |
18,15 ± 0,93 |
4,2 ± 1,0 |
93,8 |
|
2 |
2a |
* |
54,0 ± 2,0 |
18,84 ± 0,36 |
7,8 ± 1,8 |
– |
94,2 |
|
3 |
2b |
* |
* |
98,23 ± 0,59 |
88,9 ± 0,93 |
36,0 ± 1,6 |
11,3 |
|
4 |
2c |
88,96 ± 0,60 |
46,0 ± 1,3 |
24,6 ± 1,3 |
17,7 ± 1,0 |
– |
114,0 |
|
5 |
2d |
71,56 ± 0,98 |
20,8 ± 1,1 |
9,8 ± 1,2 |
– |
– |
186,3 |
|
6 |
2e |
25,1 ± 1,2 |
14,0 ± 2,3 |
4,0 ± 1,1 |
– |
– |
>250 |
|
7 |
2f |
68,9 ± 1,3 |
23,5 ± 2,9 |
4,8 ± 1,6 |
– |
– |
188,2 |
|
8 |
3'a |
* |
55,1 ± 1,2 |
33,48 ± 0,87 |
16,4 ± 1,8 |
5,2 ± 1,9 |
88,2 |
|
9 |
3'b |
27,7 ± 1,1 |
2,5 ± 2,5 |
– |
– |
– |
>250 |
|
10 |
3'c |
* |
* |
94,48 ± 0,69 |
83,48 ± 0,73 |
43,5 ± 1,4 |
12,4 |
|
11 |
3'd |
* |
87,9 ± 1,4 |
64,84 ± 0,77 |
53,7 ± 1,5 |
36,0 ± 1,3 |
21,9 |
|
12 |
3''e |
39,1 ± 1,7 |
17,6 ± 1,2 |
7,3 ± 1,5 |
– |
– |
>250 |
|
13 |
3''f |
93,1 ± 1,5 |
52,4 ± 1,7 |
6,3 ± 1,3 |
– |
– |
97,3 |
|
Acarbosea |
59,8 ± 1,2 |
21,2 ± 2,2 |
9,8 ± 1,4 |
3,2 ± 1,7 |
– |
214,5 | |
DISCUSSION
Synthesis of 3-methylrhodanine derivatives
First, we aimed to synthesize various benzylidene rhodanines from 3-methylrhodanine (1) using -substituted benzaldehydes. Six compounds, 2a–2f, were efficiently synthesized with yields ranging from 84 to 99% (Figure 3). The electronic properties of the substituents on the benzaldehydes play a role in their reaction performance. In the case of a non-substituent (R=H) or in the presence of an electron donating substituents (–CH, –OCH), products were obtained with good to high yields (Figure 3, entries 1–3). An electron withdrawing substituent such as –Cl, –NO, –CN was able to effectively activate the substrate benzaldehyde in the addition step of the carbonyl group, affording the highest yields of 97, 99, and 96%, respectively (Figure 3, entries 4–6).
The structure of products 2a–2f was determined using the H-NMR and C-NMR spectra. The signal of proton –CH= at δ 7.69–7.75 ppm in the H–NMR spectrum of each derivative allowed us to confirm the successful condensation between the –CH group of rhodanine and the carbonyl group. Furthermore, in the C-NMR spectra, the disappearance of the high field signal of CH rhodanine and the presence of a signal at δ 133.4–148.2 ppm, attributed to carbon –CH= linked with the rhodanine ring, were further supportive of this structure.
In the next step, we explored the reaction of the benzylidene rhodanines 2a–2f with phenylhydrazine. The literature12 shows that the reaction between , -unsaturated ketones and a difunctional nucleophile such as phenylhydrazine could cyclize to produce pyrazoline. Unfortunately, the results obtained confirmed that pyrazoline derivatives 3a–3f were not detected (Figure 4). However, in our case, phenylhydrazone 3'a–3'd (yields ranging from 8-14%) and pyrazole 3''e–3''f (yields of 5-7%) derivatives were formed (Figure 4).
A possible reaction mechanism for the formation of phenylhydrazone and pyrazoline has been proposed as shown in Figure 5. First, the phenylhydrazine attacks the carbonyl group, followed by dehydration to form phenylhydrazone. The cyclization reaction occurs to form pyrazoline compounds. The obtained results can be explained by the effect of the substituent on the aromatic ring. The substrates with the substituents, which weakly attract electrons to the aromatic ring (-Cl) or with the electron donating substituents (-CH, -OCH), are not able to form a pyrazoline ring. Their production stopped at the phenylhydrazone step (3’a–3’d). The substrates bearing substituents that strongly attract electrons to the aromatic ring (-NO2, -CN) can activate the C=C double bond that leads to cyclization. Hence, the electron on the nitrogen atom will attack the C=C to form the pyrazoline ring.
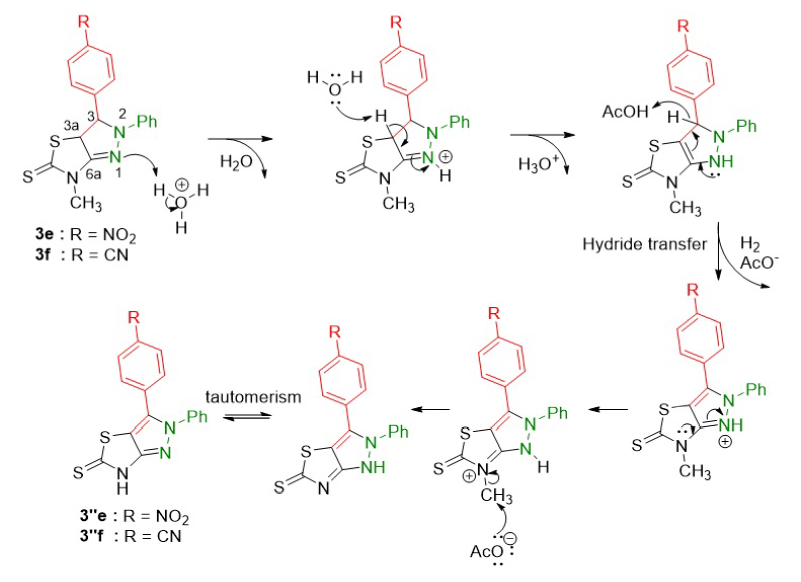
The pyrazoline derivatives 3e and 3f obtained were able to transform into pyrazole products 3''e and 3''f through oxidative aromatization (Figure 6). The process was initiated by the protonation of the nitrogen atom (N) of the pyrazoline ring. The double bond (C=N) in the pyrazoline ring then donated its electrons to the protonated nitrogen, followed by losing a hydrogen atom at C3a to form a double bond (C=C). The aromatization occurred through the hydride transfer to form a pyrazole ring. The lone pair electrons of the nitrogen N1, through resonance, are able to interact with the anti-bonding orbital of the C–H bonds and facilitate hydride transfer leading to H releasing from the intermediate to yield the desired products. The conditions of the reaction could also cleave the N-methyl group to form products 3''e and 3''f.

Proposed reaction mechanism of the synthesis of pyrazoline 3e and 3f.

Proposed reaction mechanism of the oxidative aromatization of pyrazoline to pyrazole.
Antidiabetic activity
All synthesized compounds were tested for their -glucosidase inhibitory activity. Acarbose, which is currently used clinically in combination with either diet or antidiabetic agents to control the blood glucose level of patients, was used as the positive control in this study. The IC values of these compounds are shown in
The substituents at the -position on the phenyl ring of benzylidene rhodanines 2a–2f also affected the inhibitory potential against the enzyme -glucosidase. In comparison to compound 2a (
CONCLUSION
Using a convenient and simple method, starting from 3-methylrhodanine, we have synthesized 12 derivatives in two steps: the products of the first step are benzylidene rhodanines 2a–2f with yields in the range of 84 to 99%. The products of the second step are phenylhydrazones 3'a–3'd and pyrazoles 3''e–3''f with yields from 5 to 14%. Their chemical structures were elucidated through NMR and HR-MS spectral analysis. All compounds of the second step (3'a–3'd and 3''e–3''f) are new compounds reported for the first time. As a result, the substituent on the aromatic ring showed an impressive effect in the second step. In fact, when the substrates have an electron‐donating group (-CH, -OCH) or a mild electron‐withdrawing group (-Cl) as substituent on the aromatic ring, their products stopped at the phenylhydrazone step (3'a–3'd). Otherwise, the substrates bearing substituents that strongly attract electrons to the aromatic ring (-NO, -CN) can lead to cyclization.
Moreover, the bioactivity tests showed that most of the synthesized compounds in the study had inhibitory reactions against the enzyme -glucosidase. Among them, compounds 2b, 3'c and 3'd showed the highest antidiabetic activity with IC values ranging from 11.3 to 21.9 µM. This result demonstrates that the 3-methylrhodanine derivatives have a promising application in drug development.
ABBREVIATIONS
H-NMR: Proton Nuclear Magnetic Resonance
C-NMR: Carbon-13 Nuclear Magnetic Resonance
HR-MS: High resolution - Mass spectrometry
ESI- Electrospray ionization
IC: half-maximal inhibitory concentration
TLC: thin layer chromatography
s: singlet
d: doublet
dd: doublet of doublets
t: triplet
tt: triplet of triplets
m: multiplet
ACKNOWLEDGEMENTS
This research is funded by Faculty of Chemistry, University of Science, Vietnam National University – Ho Chi Minh City under grant number HH 2021-13. We would like to thank Prof. Fritz Duus, Department of Science, Systems and Models, Roskilde University, Denmark for the gift of 3-methylrhodanine.
Authors' Contributions
Thi Hong Van Nguyen performed the experiments and interpreted the NMR and MS data. Thi Thuy Duong Ngo and Ngoc Vinh Huynh collected the biblioraphy and analyzed the experimental data of the synthesis part. That Quang Ton and Kim Phi Phung Nguyen analyzed the experimental data of the biological activities and took part of the discussion. Tan Tai Nguyen designed the research, wrote the first draft, reviewed and edited the manuscript and supervised the investigation. All the authors have read and approved the content of the final manuscript.
COMPETING INTERESTS
The author(s) declare that they have no competing interests.