

The relationship between autophagy and multidrug resistance in cancer stem cells
- Laboratory of Stem Cell Research and Application, University of Science, Ho Chi Minh City, Viet Nam
- Vietnam National University, Ho Chi Minh City, Viet Nam
- Stem Cell Institute, University of Science, Ho Chi Minh City, Viet Nam
- Laboratory of Cancer Research, University of Science, Ho Chi Minh City, Viet Nam
Abstract
Cancer stem cells (CSCs) are considered the origin of tumors and cancer. Recently, CSCs have been described as the cause of multidrug resistance (MDR) in almost all cancers. The MDR phenotype of CSCs manifests as the upregulation of ATP-binding cassette subfamily G, isoform 2 protein (ABCG2) in the cell membranes of CSCs. However, recent studies have demonstrated a relationship between MDR and the autophagy process of CSCs. Based on publications indexed in PubMed, Google Scholar, and Scopus, this review summarizes the relationship between autophagy and MDR in CSCs and the approaches to targeting autophagy to reduce MDR in CSCs. Autophagy can be considered a new target to overcome MDR in cancer treatment.
Introduction
In recent years, cancer stem cells (CSCs) have been identified as the main cause of tumor initiation, growth, metastasis, and recurrence1, 2, 3, 4, 5. Therapies aimed at targeting and eliminating CSCs have been developed 6, 7, 8, 9, 10, 11; however, the effect of these therapies remains controversial 12. To address these inconsistencies, certain research fields have been promoted to elucidate the characteristics and capabilities of CSCs 13, 14.
One of the research directions of interest is the resistance of CSCs to therapeutic agents, especially their resistance to chemotherapy15, 16. Multidrug resistance (MDR) of CSCs involves the autophagy process, which responds to stress conditions and maintains cell survival. Research has demonstrated that autophagy plays a pivotal role in the chemoresistance of various cancer cell lines 17, 18, 19, 20, 21, 22, 23.
However, observations of the correlation between autophagy and MDR in CSCs are limited. Therefore, this review aims to provide reliable evidence for elucidating the close relationship between autophagy and MDR in CSCs and suggests a promising therapy to combine chemotherapy with autophagy regulation.
Cancer stem cells
History of CSCs
The history of CSCs began in the first half of the 19 century and has undergone many stages of development over nearly two centuries to the present day 24. In the 19 century, Johannes Muller described cancer as an abnormal proliferation of “embryonic cells” that were residual and unused during development25. This idea was consolidated in the theory of cancer origin from “embryonal cell rests” that was pioneered by his pupils, Rudolf Virchow and Julius Cohnheim26. Based on this theory, a model of tumor initiation from a small group of undifferentiated cells gradually emerged.
However, it was not until the middle of the 20century that evidence of stem cells in cancer began to receive attention from scientists. In the 1950s, Leroy Stevens and Clarence C. Little demonstrated that both teratomas and teratocarcinomas were generated from highly undifferentiated cells, which were subsequently called “pluripotent embryonic stem cells” 25, 27. In 1961, Southam and Brunschwig demonstrated that only a minority population of cancer cells derived from patients had tumorigenic capacity when autotransplanted to different sites 6, 28. In 1963, Bruce et al. emphasized the pivotal role of a small group of lymphoma cells in tumor initiation 29. In 1964, Kleinsmith and Pierce further demonstrated that embryonal carcinoma (EC) cells isolated from cancer tissue had diverse differentiation potentials 27. In 1971, Pierce published evidence that differentiated cancer cells could not form tumors when injected into experimental mice30. The results indicated that tumor initiation and development were facilitated by a small group of undifferentiated cancer cells that were highly proliferative and had multidifferentiation potential. This was the foundational basis of the concept of CSCs.
In the 1990s, studies on human acute myeloid leukemia (AML) cells by Lapidot (1994) and Bonnet and Dick (1997) indicated that only a subpopulation of cells expressing specific surface markers CD34/CD38 acted as the initiating cells in tumors30, 14. Through this evidence, CSCs were officially identified and isolated from the cancer cell population. Subsequent studies showed that tumor-initiating cells, also known as CSCs, are characterized by distinct markers for different cancer tissues14. To date, numerous CSCs have been isolated and enriched due to their specific cell markers. An increasing number of studies have targeted CSCs to improve the effect of cancer therapies.
Characteristics of CSCs
Based on the results of existing studies, previous reports have proposed the characteristics of CSCs that contribute to their role in tumor initiation, survival, and development. These include (1) tumorigenesis capability, (2) self-renewal and differentiation into multiple cell lines, (3) expression of specific markers for isolation, (4) maintenance of a “stemness property” after more transplanted generations, and (5) resistance to conventional therapies31, 32, 33.
The tumor-forming ability of CSCs involves their cell origin. CSCs originate from normal stem cells or progenitor cells acquiring “stem cell attributes” 34, 35, 36, 37. By stimulating the microenvironment, these cells undergo uncontrolled proliferation and transform into CSCs 38, 39. Therefore, CSCs are inherited, self-renewing, multilineage-differentiated stem cells that are capable of driving tumor development. Similar to normal stem cells, the self-renewal capability of CSCs is regulated by specific signaling pathways, such as the Wnt/β-catenin, Notch, and Hedgehog pathways 40, 41, 42, 43. In addition, the other pathways of tumor suppressor genes, represented by phosphatase and tensin homolog on chromosome 10 (PTEN) and tumor protein p53 (TP53), contribute to both self-renewal and tumor initiation in CSCs 44. Furthermore, the self-renewal and multidifferentiation potential of CSCs results in a hierarchy population of cancer cells that explains the existence of heterogeneous tumors 45, 46. The self-renewal characteristic of CSCs is the main basis for maintenance of their tumorigenesis potential during serial transplantations in subsequent mouse generations. Currently, the transplantation assay is used to identify the hallmarks of CSCs 47, 48, 49, 50, 51.
Another characteristic of CSCs is the expression of specific markers (i.e., cell membrane receptor proteins) on the cell surface. The difference in cell surface markers between CSCs and other tumor cells suggests a method for isolating CSCs from the cancer cell population52, 53, 54, 55. CSCs that are isolated from different tissues express groups of distinctive molecular markers 31, 33, such as CD44/CD24/ESAin pancreatic CSCs 56, 57, ESA/CD44/CD24/Linin breast CSCs58, and CD133/α2β1 and integrin/CD44markers in prostate CSCs59. Due to the specificity of these markers in CSCs, they have been proposed as potential targets for cancer therapies60.
Although targeting CSCs seems promising to improve tumor treatment efficiency, it encounters an inherent problem that involves the resistance of CSCs to a majority of commonly used treatments. In general, the anti-therapy mechanisms of CSCs are divided into two types of resistance: acquired and intrinsic61. Acquired resistance is based on the response of the CSCs to therapeutic agents. Radiotherapy not only stimulates the DNA damage checkpoint and activates the DNA-repair systems of CSCs62, but it also activates the defense mechanism against reactive oxygen species (ROS) 63. In contrast, chemotherapy is both influenced by acquired resistance, like radiotherapy, and affected by the intrinsic resistance of CSCs through processes such as quiescence (or dormancy), self-renewal, transformation between cell phenotypes (or plasticity), and the expression of drug transporters and detoxification proteins. In addition, the antitherapeutic activity of CSCs is supported by interaction between the tumor microenvironment and CSCs to generate resistance through signaling pathways 64. Understanding of the mechanisms of CSC resistance paves the way for novel cancer treatment strategies that focus on inhibiting these mechanisms and reversing the sensitivity of CSCs to therapeutic agents65.
Multidrug resistance of CSCs
Antichemotherapeutic activity of CSCs
One of the most popular cancer therapies is chemotherapy. Numerous drugs that are efficient in inducing cell death have been used to treat a variety of cancers 66, 67. MDR is defined as the “simultaneous resistance of cancer cells toward a broad spectrum of structurally unrelated cytotoxic drugs that have different modes of action” 68. There are two types of chemoresistance in tumors: primary (or de novo) resistance and acquired resistance, which can be observed in ovarian cancer 69, glioblastoma68, 70, 71, pancreatic cancer72, breast cancer73, neuroblastoma, and hepatoblastoma 66. Primary resistance (also called intrinsic resistance) confers drug resistance via factors that are intrinsic to cancer cells in tumors, usually due to CSC aptitude, before the administration of chemotherapies. Acquired resistance (or extrinsic resistance) is the acquired ability formed by the responsiveness of cancer cells to chemotherapy via genetic and epigenetic modifications for detoxification68, 69, 74.
Mechanisms of MDR in CSCs
Recent insights into CSCs have indicated their essential roles in MDR. Investigation of MDR mechanisms in CSCs provides an opportunity to overcome them 75, 76, 77. MDR of CSCs is based on many cellular activities, such as the DNA repair system, transporter efflux pump, detoxification enzymes (aldehyde dehydrogenase, DNA topoisomerase, protein kinase C, dihydrofolate reductase, glutathione and glutathione S-transferases [GST]), EMT, autophagy, oncogenes (EGFR, PI3K/AKT, ERK, and NF-кB), microRNAs, tumor suppressor genes (e.g., p53), and B-cell lymphoma 2 (Bcl-2). In addition, microenvironmental conditions, such as hypoxia, pH, and paracrine signals, affect the drug-resistance capacity of CSCs78, 79, 80, 81, 82, 83.
Protein activity plays a role in the form of efflux pumps that excrete a broad range of chemotherapeutic drugs (e.g., doxorubicin [DOX], cisplatin, 5-fluorouracil [5-FU], colchicine, methotrexate, etoposide) out of CSCs, thereby preventing their cytotoxicity and supporting the chemoresistance of CSCs82. A main protein family for this task is ATP-binding cassette (ABC) transporters. Their crucial function is to transport a variety of substances, such as peptides, inorganic anions, amino acids, polysaccharides, proteins, vitamins, and metal ions. In CSCs, they function as a system to efflux toxins. ABC transporters are divided into seven subfamilies with 49 members, named ABC-A to ABC-G. An ABC protein has four domains: two nucleotide-binding domains (NBDs) and two transmembrane domains (TMDs)84. The expression of ABC transporters is affected by the signaling pathway, and energy from the hydrolyzation of a pair of ATP molecules that bind to transporters can drive the active transport of drugs and/or other substances out of cells 85.
DNA repair systems that help to detect and repair mismatches on DNA strands are another important MDR mechanism in CSCs86. In general, DNA damage induced by both intra- and extracellular factors (e.g., endogenous ROS, ultraviolet radiation, X- and gamma rays, plant toxins, mutagenic chemicals, and chemotherapeutic agents)80 activates a response network. First, DNA errors are identified by sensor complexes, including Mre11-Rad50-Nbs1 (MRN), which recognize DNA double-strand breaks (DSBs), while the RPA-ATRIP complex recognizes single-strand breaks (SSBs). Then, the repair systems restore DNA damage via six mechanisms: (1) the direct reversal pathway (MGMT, ABH2, ABH3), (2) the mismatch repair (MMR) pathway, (3) the nucleotide excision repair (NER) pathway, (4) the base excision repair (BER) pathway, (5) the homologous recombination (HR) pathway, and (6) the nonhomologous end-joining (NHEJ) pathway87, 88.
Despite the activation of DNA repair systems, drugs still cause extensive damage. To survive, CSCs may prevent apoptosis by promoting the action of the Bcl-2 protein family, such as Bcl2-associated-X-protein (Bax), Bcl-2 homologous antagonist killer (Bak), B-cell lymphoma-extra small (Bcl-X), and anti-apoptosis proteins, such as Bcl-2, B-cell lymphoma-extra-large (Bcl-X), and myeloid cell leukemia 1 (Mcl-1) 79, 89. Under normal circumstances, apoptosis is induced by proapoptotic proteins via stimulation of apoptogenic proteins, such as cytochrome c produced by mitochondria. However, proapoptotic proteins are associated with antiapoptotic proteins that reduce their activity and interfere with cellular apoptosis90. The Bcl-2 protein family entirely restricts a variety of drugs: dexamethasone, cytosine arabinoside (Ara-C), methotrexate, cyclophosphamide, adriamycin, daunomycin, S-fluoro-deoxy-uridine, 2-chlorodeoxyadenosine, fludarabine, paclitaxel (Taxol), etoposide (VP-16), camptothecin, nitrogen mustards, mitoxantrone, cisplatin, vincristine, and some retinoids. Although these drugs affect different pathways, antiapoptotic Bcl-2 and its family members impede the effectiveness of drugs by inhibiting signals inducing cell death. Therefore, even if toxic molecules can penetrate the cell and destroy the DNA structure, cancer cells still survive and are able to prevent the effects of the drugs, repair DNA damage, and proliferate91.
Another MDR mechanism of CSCs involves a group of enzymes that are essential components in metabolism pathways: drug metabolism enzymes (DMEs). There are two phases of DMEs distinguished by two basic but distinct reactions. Phase I enzymes or metabolism enzymes comprise cytochrome P450 enzymes (CYPs), oxidoreductases, and epoxide hydrolases that oxidize appropriate substrates, namely therapeutic drugs. This oxidation alters the substrates to form favorable molecular structures for activating enzymes in phase II. Subsequently, phase II enzymes or transferase enzymes, such as GSTs, UDP glucuronosyltransferases (UGTs), sulfotransferases, and arylamine N-acetyltransferases (NATs), conjugate definite complexes to targeted substrates to create nontoxic compounds88, 92, 93, 94. For example, UGTs have been demonstrated to transfer the glucuronic acid component of UDP-glucuronic acid to anthracycline (daunorubicin), which correlates with a reduction in daunorubicin cytotoxicity 95. Finally, these compounds are pumped out of cells via ABC transporters 88.
The conditions of the microenvironment, such as hypoxia or low pH, also contribute to hindering drug efficacy. Under hypoxic conditions, the low oxygen concentration reduces the cytotoxicity of chemotherapeutic drugs due to their oxidation requirement to transform into cytotoxic structures 88. In addition, the hypoxia-inducible factors (HIFs) produced in response to hypoxia induce the expression of ABC transporters78. Furthermore, hypoxic conditions facilitate intracellular accumulation of lactic acid via the glycolysis pathway. Therefore, cancer cells induce proton pumps to efflux Hions into the extracellular space and promote acidification of the microenvironment. The high concentration of extracellular Hions causes “ion trapping,” ionizing weak bases to become positively-charged complexes. Because of the ion trapping phenomenon, the cell permeability of weakly basic chemotherapeutic agents (e.g., DOX, mitoxantrone, vincristine, anthracyclines, anthraquinones, vinca alkaloids) is decreased, and the effects of the drugs are impaired96, 97. Other components of the tumor microenvironment, including the extracellular matrix (ECM), matrix rigidity, hypervascularization, and paracrine factors, mediate chemoresistance by controlling drug availability, stimulating EMT, and promoting oncogenic signaling pathways.
Autophagy of CSCs
Discovery and definition
Autophagy is a combination of two words that originate from Greek: “auto” means self, while “phagy” means eating, so autophagy means “self-eating” 98. This process was discovered many years ago. In 1859, this term was first introduced under the name “autophagie” in a magazine published by the French Academy of Science and was used by physiologist M. Anselmier 99. In 1963, Christian de Duve was the first to use the term autophagy in accordance with its current functional definition: a process by which cellular materials are taken to and decomposed in lysosomes (in animals) or vacuoles (in plants, yeasts)100. To date, 42 autophagy-related genes (ATGs) responsible for autophagosome formation and autophagy regulation have been identified101. Microtubule-associated protein 1 light chain 3 (LC3), which is the main autophagy indicator in mammals, was identified by Kabeya et al.102. Beclin 1 was reported to play dual roles as an autophagy inducer when it binds to phosphatidylinositol 3-kinase 103 and as a tumor suppressor due to its mediation of E-cadherin localization 104, 105.
In summary, many previous reports have indicated that autophagy is an important cellular process through which different cytoplasmic components are broken down and recycled via lysosomal degradation106. This process is often activated in response to a shortage of nutrients, leading to regeneration of other organelles and substances to provide essential precursors for metabolic activity 107.
In vitro and in vivo mechanisms
There are three main types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) 100. Macroautophagy (hereafter termed “autophagy”) is the most studied form. Macroautophagy mainly involves the degradation of long-lived proteins via lysosomes108, especially faulty proteins in specific diseases, such as huntingtin (in Huntington’s disease109), a-synuclein (in Parkinson’s disease110), or fibrinogen g-chain (in hypofibrinogenemia111). After receiving a stress signal, macroautophagy begins in the cytoplasm with the formation of a double-membrane-bound structure called an autophagosome112. Autophagosomes then fuse with lysosomes to form autolysosomes, where their cytoplasmic contents are degraded by hydrolases and sent back to the cytoplasm as recycling material for cellular metabolism 112. In microautophagy, the membranes of lysosomes or vacuoles are randomly invaded and differentiate into autophagic tubes enclosing cytosolic components 113, which are then degraded by hydrolases 100.
The two phenomena mentioned above were described by Duve and Wattiaux in rats in 1966 113. Fifteen years later, CMA was first described in human fibroblasts cultured in medium without serum containing growth factors 114. The CMA process begins when degraded proteins are recognized by cytosolic chaperone complexes and brought to the surface of lysosomes. At the lysosomal membrane, after binding to specific molecules, the proteins are translocated into the lumens of lysosomes, where they are completely degraded 115. CMA is known to be activated as part of the amino acid response during prolonged nutritional starvation116.
Recently discovered types of autophagy with roles in specific organelles are being studied to elucidate their mechanisms and functions. Typical examples include mitophagy (mediating mitochondria removal) 117, ribophagy (degradation system for ribosomes)118, xenophagy 119, and lipophagy (connection between autophagy and lipid metabolism) 120.
Relationship between autophagy and MDR in CSCs
Autophagy contributes to MDR in CSCs
The contribution of autophagy to MDR development in CSC lines has been investigated for many years. An underlying mechanism of autophagy-stimulated MDR in CSCs has also been gradually elucidated. In 2013, Wu et al. evaluated the resistance of colon CSCs, which were isolated from the SW1222 and HCT116 cell lines via CD44/CD24 markers, to paclitaxel. A cytotoxic result indicated that SW1222 stem cells were more resistant to paclitaxel than HCT116 stem cells. A further experiment on signaling pathways demonstrated that Cdx1 stimulated autophagy activation by increasing Bcl-2 and LC3-II levels in SW1222 stem cells. In addition, silencing Cdx1 expression with siRNAs or inhibiting autophagy with the lysosomal inhibitor bafilomycin A250 (BafA) caused SW1222 stem cells to become more sensitive to paclitaxel. However, HCT116 stem cells do not express Cdx1 but express p53, which induces apoptosis due to increased expression of the Bax protein. Reviving the expression of Cdx1 in HCT116 stem cells promotes autophagy, significantly restricting apoptosis in these cells 121. In conclusion, Cdx1-induced autophagy based on the Cdx1-Bcl-2-LC3-II signaling pathway plays a pivotal role in the resistance of colon CSCs to paclitaxel.
Another study on colorectal cancer (CRC) was published by Yang et al. in 2015. The role of autophagy in chemoresistance was investigated in both CRC cells and CSCs. In the CRC cell lines SW620 and SW480, autophagy is induced by oxaliplatin; otherwise, the hypoxia/starving (H/S) environment enhances autophagy activation in CRC cells. The results indicated that autophagy reduced cell death by inhibiting oxaliplatin-induced apoptosis in CRC cells cultured in an H/S environment. In addition, treatment with oxaliplatin was demonstrated to enrich CD44 CRC cells, especially when they were cultured in an H/S environment. In further investigations, the enriched CD44 CRC cells were sorted to obtain CSCs based on CD44, which is a characteristic surface marker for colon cancer CSCs. CSCs (CD44 cells) and CD44 cells were exposed to oxaliplatin. The data indicated that autophagy enhanced by the H/S environment improved the survival proportion of CD44 cells, which was higher than that of CD44 cells. In contrast, the presence of 3-methyladenine (3-MA) prevented autophagy activation so that the survival of both cell groups was not significantly different 122. Therefore, autophagy stimulated by stress conditions functions to inhibit the oxaliplatin effect and promotes the survival of colorectal CSCs.
In another study, Yue You et al. demonstrated that autophagy regulated by BCRA1 enhanced the drug resistance of ovarian CSCs to cisplatin. BCRA1 is a tumor suppressor gene that contributes to multiple cell processes in cancerous tissues, especially drug resistance. The results revealed that SKOV3 cells, an ovarian cancer cell line, inhibited increased expression of both autophagy proteins and BCRA1. SKOV3 cells were isolated from epithelial ovarian cancer stem cells (EOCSCs) via CD133. Comparison of EOCSCs and parental cells revealed that EOCSCs had higher activation of autophagy and BCRA1 than SKOV3 cells, which had a greater effect on stemness and drug resistance. Furthermore, transfection of the BCRA1 plasmid into EOCSCs resulted in overexpression of BCRA1 and upregulation of Beclin‐1, ATG5, P‐gp, and ABCG2. The increase in the LC3-II/I ratio confirmed the regulation of autophagy by BCRA1. Furthermore, knocking down BCRA1 and inhibiting autophagy sensitized EOCSCs to cisplatin due to increased apoptosis and interference with the cell cycle. In addition, treatment with torkinib stimulated autophagy and attenuated cell cycle arrest in EOCSCs with BRCA1 knockdown123. The results indicate crosstalk between BCRA1 and autophagy that allows BCRA1 to increase cisplatin resistance in EOCSCs through autophagy.
A recent report by Li et al. suggested a relationship between autophagy and chemoresistance in gastric CSCs. Stem cells were isolated from the gastric cancer cell lines MGC-803 and MKN-45 using CD54 and CD44 markers. The LC3-II proportion was enhanced in CD44/CD54 gastric cells to induce increased activity of autophagy in these cells compared with the original cancer cells. Treatment of gastric CSCs with a combination of chloroquine (CQ) and 5-FU revealed that autophagy inhibition increased the chemosensitivity of gastric CSCs. Furthermore, suppressing the Notch signaling pathway enhanced cell death in chemotherapy-treated gastric CSCs. This result indicated an association of the Notch signaling pathway with autophagy-mediated chemoresistance in gastric CSCs124. This evidence indicates the effect of autophagy on chemoresistance in gastric CSCs based on the Notch signaling pathway.
The described studies investigated the effective mechanisms of autophagy through signaling pathways to regulate MDR in CSCs from different tissues. These results consolidate an essential contribution of autophagy to chemoresistance in CSCs.
Autophagy facilitates reversal of MDR in CSCs
Autophagy has been declared a “double-edged sword”125, 126. In brief, this process not only plays a pro-survival role to protecting cancer cells from therapeutic drugs, but it also kills resistant cells by stimulating programmed cell death and facilitating MDR reversal23. Although the mechanism of autophagy-mediated cell death in cancer cells, especially CSCs, is unclear, some evidence indicates that autophagy can induce cell death of MDR cells 127, 128, 129 or promote apoptotic signaling pathways 23.
Some studies on rottlerin, a plant-derived chemopreventive agent isolated from Mallotus philippensis 130, 131, reported that autophagy induced by rottlerin is followed by induction of apoptosis132. CSCs treated with rottlerin exhibited enhanced conversion of LC3-I to LC3-II, which is a hallmark of autophagy 131. In addition, rottlerin increased the expression of Atg7 and Beclin-1 in pancreatic CSCs132, Atg12 in breast CSCs130, and other genes in prostate CSCs131, which accumulated during autophagy processing. In contrast, silencing of the Atg7 and Beclin-1 genes led to the inhibition of rottlerin-induced autophagy132. Furthermore, rottlerin-treated CSCs suppressed the phosphorylation of the PI3K/AKT/MTOR signaling pathway, which is associated with the maintenance of CSCs 133, decreased expression of anti-apoptosis proteins, such as Bcl-2, Bcl-XL, XIAP, and cIAP-1, induction of Bax, activation of caspase-3 and -9, and concomitant degradation of poly (ADP-ribose) polymerase (PARP). These results confirmed the correlation between rottlerin treatment and apoptosis induction. Moreover, there are also data indicating that the inhibition of autophagy by 3-MA and bafilomycin may arrest apoptosis130. In summary, rottlerin-induced autophagy mediates apoptosis in CSCs from different tumors via inhibition of the PI3K/AKT/mTOR signaling pathway.
Another report in MDR human A549 lung cancer cells by Kaewpiboon et al. demonstrated that feroniellin A (FERO) reduces the expression of NF-κB, which correlates with MDR reversal and leads to sensitization to apoptosis via downregulation of P-gp. In addition, FERO enhances the conversion of LC3-I to LC3-II and induces autophagy, and the activation of autophagy by rapamycin increases FERO-induced apoptosis. This evidence suggests that FERO-induced autophagy functions as a mediating factor in reversing MDR and facilitating apoptosis in MDR human A549 lung cancer cells134. Furthermore, Xu et al. reported that cryptotanshinone (CTS), an active quinoid diterpene isolated from Salvia miltiorrhiza Bunge, induces autophagic cell death in MRD colon cancer cells based on activation of the ROS-p38/MAPK/NF-kB signaling pathway 135.
These studies provide evidence that autophagy induced by the identified substances can stimulate programmed cell death and MDR reversal in both CSCs and MDR cancer cell lines in some cases.
Autophagy is a potential target to overcome the MDR of CSCs
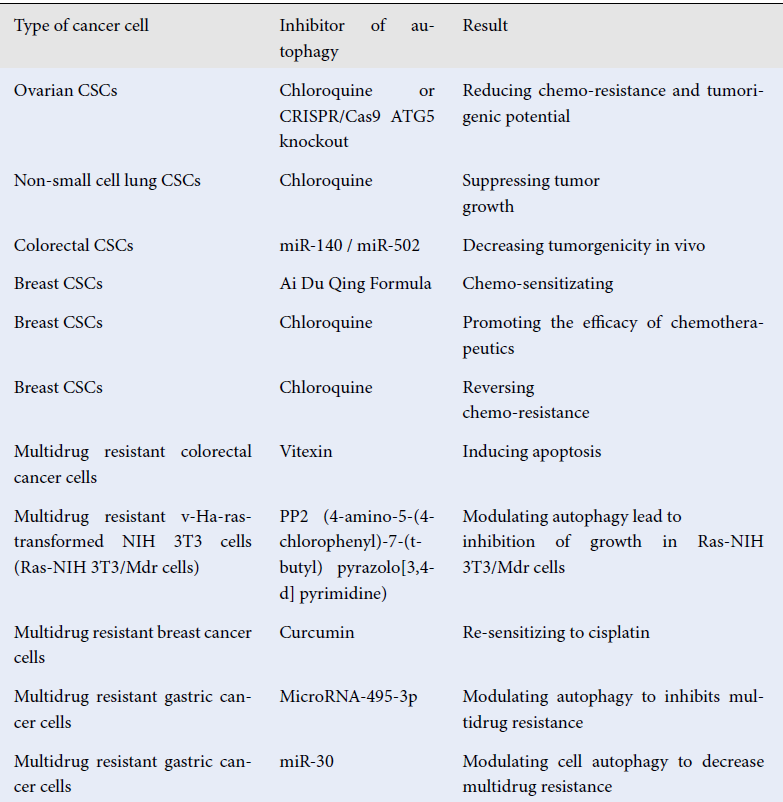
According to the previously mentioned studies, autophagy and MDR in CSCs have an intimate relationship. Therefore, autophagy has become a potential target to overcome the MDR of CSCs in the last decade. A report by Pagotto et al. indicated that inhibiting autophagy using CQ or CRISPR/Cas9 ATG5 knockout reduced both chemoresistance in vitro and tumorigenicity in vivo in human ovarian CSCs136. Another study demonstrated that autophagy suppression by CQ in CSCs promoted chemosensitivity to cisplatin in non-small cell lung carcinoma137. A report on colon CSCs showed that microRNAs could be utilized to disrupt autophagy to promote apoptosis, overcome MDR, and decrease the tumorigenicity of CSCs138. Furthermore, Liao et al. demonstrated that autophagy blockade by Ai Du Qing formula, a traditional Chinese medicine, attenuated the GRP78/β-Catenin/ABCG2 signaling pathway and stimulated the chemosensitivity of breast CSCs 139. In the same case, Sun et al. demonstrated that the combination of inhibiting autophagy and chemotherapy by nanoparticles loaded with CQ, DOX, and docetaxel (DTXL) increased the effect of the drug on breast CSCs140. Bousquet et al. investigated whether inhibition of the autophagic pathway of breast CSCs reverses the chemoresistance of these cells in pretreatment biopsies of triple negative breast cancer patients141. Other studies have provided additional evidence that autophagy inhibition leads to sensitization of cancer cells to drugs, apoptosis induction, and decreased resistance in MDR cancer cells (
Overcoming chemo-resistance of CSCs and multi-drug resistant cells by targeting to autophagy
|
Type of cancer cell |
Inhibitor of autophagy |
Result |
Reference |
|---|---|---|---|
|
Ovarian CSCs |
Chloroquine or CRISPR/Cas9 ATG5 knockout |
Reducing chemo-resistance and tumorigenic potential |
|
|
Non-small cell lung CSCs |
Chloroquine |
Suppressing tumor growth |
|
|
Colorectal CSCs |
miR-140 / miR-502 |
Decreasing tumorgenicity in vivo |
|
|
Breast CSCs |
Ai Du Qing Formula |
Chemo-sensitizating |
|
|
Breast CSCs |
Chloroquine |
Promoting the efficacy of chemotherapeutics |
|
|
Breast CSCs |
Chloroquine |
Reversing chemo-resistance |
|
|
Multidrug resistant colorectal cancer cells |
Vitexin |
Inducing apoptosis |
|
|
Multidrug resistant v-Ha-ras-transformed NIH 3T3 cells (Ras-NIH 3T3/Mdr cells) |
PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d] pyrimidine) |
Modulating autophagy lead to inhibition of growth in Ras-NIH 3T3/Mdr cells |
|
|
Multidrug resistant breast cancer cells |
Curcumin |
Re-sensitizing to cisplatin |
|
|
Multidrug resistant gastric cancer cells |
MicroRNA-495-3p |
Modulating autophagy to inhibits multidrug resistance |
|
|
Multidrug resistant gastric cancer cells |
miR‑30 |
Modulating cell autophagy to decrease multidrug resistance |
|
Conclusion
Evidence from scientific reports reveals an intimate correlation between MDR and autophagy in CSCs. The elucidation of this relationship will pave the way to understanding the anti-therapeutic mechanism of tumors, thereby contributing to resolving challenges in current cancer treatment.
Acknowledgments
This research is funded by Vietnam National University HoChiMinh City (VNU-HCM) under grant number C2020-18-27 and University of Science, VNU-HCM under grant number T2021-62.
Authors’ contributions
Nhan Ngo-Tran The and Khan Dinh Bui have equal role in composing the content. Phuc Van Pham suggests ideas for the manuscript, checks form of presentation and edits script.
Conflicts of interest
There are no conflicts of interest among authors.
Patient consent
Not applicable
Ethics approval
Not applicable
List of abbereviations
ABC transporter: ATP-binding cassette transporter, ALDH: aldehyde dehydrogenase, AMPK: AMP-activated protein kinase, ATG: autophagy-related gene, CD: cluster of differentiation, CMA: chaperone-mediation autophagy, CSCs: Cancer stem cells, EMT: epithelial-to-mesenchymal transition, LC3: light chain 3, MDR: Multidrug resistance, mTOR: mechanistic target of rapamycin