

Hydrophobic Property of (R)-3 Amidinophenylalanine Inhibitors Contributes to their Inhibition Constants with Thrombin Enzyme
- Department of Chemistry, Faculty of Sciences, Nong Lam University, Vietnam
- Department of Chemical Technology, Faculty of Chemical and Food Technology, Ho Chi Minh City University of Technology and Education
Abstract
Introduction: Thrombin is the key enzyme of fibrin formation in the blood coagulation cascade. Thrombin is released by the hydrolysis of prothrombinase which is generated from factor Xa and factor Va in the presence of calcium ion and phospholipid. The inhibition of thrombin is of therapeutic interest in blood clot treatment. Currently, potent thrombin inhibitors of (R)-3- amidinophenylalanine, derived from benzamidine-containing amino acid, have been developed so far. In order to quantitatively express a relationship between chemical structures and inhibition constants (Ki with thrombin enzyme in a data set of (R)-3-amidinophenylalanine inhibitors), we developed a quantitative structure-activity relationship (QSAR) modeling from a group of 60 (R)-3- amidinophenylalanine inhibitors.
Methods: A database containing chemical structures of 60 inhibitors and their Ki values was put into molecular operating environment (MOE) 2008.10 software, and the two-dimensional (2D) physicochemical descriptors were numerically calculated. After removing the irrelevant descriptors, a QSAR modeling was developed from the 2D-descriptors and Ki values by using the partial least squares (PLS) regression method.
Results: The results showed that the hydrophobic property, reflected through n-octanol/water partition coefficient (P) of a drug molecule, contributes mainly to Ki values with thrombin.The statistic parameters that give the information about the goodness of fit of a 2D-QSAR model (such as squared correlation coefficient of R2 = 0.791, root mean square error (RMSE) = 0.443, cross-validated Q2 cv = 0.762, and cross-validated RMSEcv = 0.473) were statistically obtained for a training set (60 inhibitors). The R2 and RMSE values were obtained by using a developed model for the testing set (9 inhibitors) ; the total set has statistically significant parameters. Furthermore, the 2D-QSAR modeling was also applied to predict the Ki values of the 69 inhibitors. A linear relationship was found between the experimental and predicted pKi values of the inhibitors.
Conclusion: The results support the promising application of established 2D-QSAR modeling in the prediction and design of new (R)-3-amidinophenylalanine candidates in the pharmaceutical industry.
Introduction
Fibrin clot formation is an important process that heals a wound and stops any unwanted bleeding. However, an abnormal clot in the bloodstream leads to pain and swelling because the blood gathers behind the clot. As a result, a heart attack can occur. There are pathways (mechanisms) which lead to fibrin formation. The intrinsic pathway was proposed in which fibrin formation resulted from a series of stepwise reactions involving only proteins circulating in blood as precursors or inactive forms 1,2,3. Proteins were activated by proteolytic reactions and converted to thrombin. The intrinsic mechanism can be triggered when thrombin is generated, leading to the activation of factor XI 2. The extrinsic pathway requires tissue factor VII in blood 2,3,4,5. Initially, a complex including factor VII was formed via calcium ion dependent reaction and then converted factor VII to factor VIIa (a: activated). The activation of many factors, including factor V, VIII, IX and X, in sequence results in the generation and release of thrombin. When thrombin is formed, it converts fibrinogen to fibrin by proteolysis. Finally, the cross-linking reactions were catalyzed by an activated factor XIIIa to form a very strong fibrin clot 2.
As discussed above, thrombin is a key enzyme in fibrin formation. Therefore, inhibitors selective toward thrombin have been developed; these include peptide aldehydes6 and boronic acid derivatives7. The anticoagulants derived from 3-amidinophenylalanine that are associated with their inhibition constants (K values) toward thrombin enzyme have been reported 8,9. The inhibition constant is an equilibrium constant of the reversible combination of the enzyme with a competitive inhibitor, I + E <->IE (K = [IE]/[I][E] ([I], [E] and [IE] are the equilibrium concentrations of inhibitor (I), enzyme (E), and enzyme-inhibitor complex (IE))10. The K value reflects the binding affinity of drug to target. The greater the binding affinity, the larger the K value is, i.e., the less amount of medication needed to inhibit the enzyme.
The design and synthesis of thrombin inhibitors could be improved in several ways. The two dimensional-quantitative structure-activity relationship (2D-QSAR) is one of the insilico drug discovery approaches due to its reliability and interpretability. In principle, the 2D-QSAR can be used to extract physicochemical properties (descriptors) which mainly contribute to the bioactivity of drug candidates 11. In the present work, in order to express the 2D-descriptors playing a crucial role on K of a series of (R)-3-amidinophenylalanine inhibitors, we applied 2D-QSAR method to develop a mathematical QSAR equation from 60 inhibitors as a training set. The modeling was then used to predict K values of 69 inhibitors toward thrombin enzyme.
Methods
A data set of 69 inhibitors derived from (R)- 3-amidinophenylalanine and their logarithm of inhibition constants, pK = - logK toward thrombin enzyme was selected for the 2D-QSAR study 8 (Figure 1). Chemical structures of inhibitors were drawn in molecular operating environment (MOE) 2008.10 software and then optimized energetically prior to doing calculations. In order to develop a mathematical 2D-QSAR model, a training set containing 60 inhibitors was randomly selected in MOE. The selection of a training set was done when all parameters such as squared correlation coefficient (R), cross-validated correlation coefficient of Qand root-mean-square error (RMSE) of internal and external validations were statistically significant. In our study, this was repeated 8 times to obtain a satisfied training set. The remaining 9 inhibitors were used as a testing set to evaluate the reliability of the model. The input data were chemical structures and pK values of inhibitors. The 2D-molecular physicochemical properties (descriptors) are numerical values and calculated by using MOE. The inhibition constants, K, depended on 184 2D-molecular descriptors. However, the irrelevant descriptors which showed a zero value, a low correlation (< 0.07) with K,and high intercorrelation (> 0.7) between themselves were discarded. These descriptors were screened out using the Rapidminer 5 software. In addition, QuaSAR-Contigency and Principle Components in MOE 2008.10 were also used to screen the most relevant descriptors. The partial least squares (PLS) regression method was used to develop a 2D-QSAR model. This model was used to predict the K values of 69 inhibitors and were predicted via the QuaSAR Fit validation panel in MOE.
Results
2D-QSAR modeling
The first goal of this work is to develop a 2D-QSAR modeling which presents molecular descriptors of (R)-3-amidinophenylalanine inhibitors which predominantly contribute to the inhibition constant, K. The selected 2D-QSAR equation is given below:
Here, SlogP_VSA0, SlogP_VSA1, SlogP_VSA3 are molecular descriptors associated with coefficients. The training set was randomly selected, we have analyzed to develop significant models by using different training set with additional descriptors. The goal was to explain and search for other descriptors that relate to the inhibition constant. Unfortunately, other developed models possessed poor R, Q and RMSE parameters. Therefore, those models could not be used for further analysis and discussion.

Chemical structures, experimental (Exp) pKi
Statistical parameters
The statistical parameters (such as R, Q and RMSE) give information about the goodness of fit of a model. The best model is selected when it possesses highest R values, Q (> 0.5) values, and lowest RMSE (< 0.5)11.
Predicted pK values using a developed 2D-QSAR model
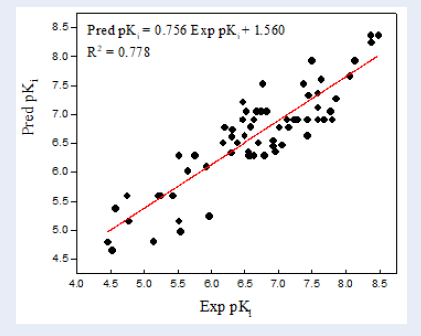
Lastly, the pK values of 69 inhibitors were predicted using the established 2D-QSAR modeling. The pK values of all molecules are listed in Figure 1. A plot of experimental vs. predicted pK is shown in Figure 2.

The plot of correlations representing the experimental
Statistically significant parameters of the established 2D-QSAR model
| Training set | Cross-validation | Testing set | ||
|---|---|---|---|---|
| No | 60 | 60 | 9 | 69 |
| R2 | 0.791 | 0.962 | 0.771 | |
| Q2cv | 0.762 | |||
| RMSE | 0.443 | 0.473 | 0.161 | 0.460 |
Discussion
Molecular descriptors
By using the partial least squares regression method, a 2D-QSAR modeling was established from a data composing of numerically relevant descriptors and pK values of 60 inhibitors with thrombin. The developed modeling expressed the dependence of pK on the hydrophobic descriptor. The logP refers to logarithm of the n-octanol/water partition coefficient (P). This property is an atomic contribution model that calculates logP from the given structure 12. This descriptor was used as a measure of cell permeability of the drug molecule. The partition coefficient is a ratio between the concentrations of a solute in lipid phase (n-octanol) and in aqueous phase (P = C/C. Compounds possessing P > 1 are lipophilic or hydrophobic while compounds for which P < 1 are hydrophilic. LogP of a molecule was calculated from fragmental or atomic contributions (surface area, molecular properties, and solvatochromic parameters) and various correction factors (electronic, steric, or hydrogen-bonding effects)11,13. Each atom has an accessible van der Waals surface area (VSA), a, along with an atomic property, p. This property is in a specified range (a, b) and contributes to the descriptor. Slog P_VSA is the sum of a of all atoms, such that p value of each atom i is in a range of (a, b) (
Molecular descriptors in 2D-QSAR modeling
| Descriptor Code | Description |
|---|---|
| SlogP_VSA0 | Sum of ai such that pi <= -0.4 |
| SlogP_VSA1 | Sum of ai such that pi is in (-0.4, -0.2] |
| SlogP_VSA3 | Sum of ai such that pi is in (0.0, 0.1] |
2D-QSAR modeling and its validation
The selected 2D-QSAR modeling is a model possessing statistically significant parameters of internal and external validations. The developed model (
Conclusions
The 2D-QSAR modeling has been successfully developed from 2D-descriptors of 60 (R)-3-amidinophenylalanine inhibitors associated with their inhibition constants, K. The established QSAR modeling was internally, externally, and totally validated, demonstrating satisfactory statistical parameters. Hydrophobicity is an important descriptor in the modelling of binding affinity. The 2D-QSAR equation was applied to predict K values of all inhibitors. The results revealed a good predictability of the modeling. Based on the developed 2D-QSAR modeling, the design of the new inhibitors derived from (R)-3-amidinophenylalanine should focus on the hydrophobicity of derivatives by theoretical calculations to obtain the numerical values of hydrophobic descriptors. The chemical structures of inhibitors possessing lower values of SlogP_VSA1, SlogP_VSA3 descriptors and higher SlogP_VSA0 descriptor should be further studied in synthetic experiments.
List of abbreviations
2D-QSAR: two dimensional-quantitative structure-activity relationship
CV: cross-validation
LOO: leave one out
MOE: molecular operating environment
RMSE: root-mean-square error
Author Contributions
The contributions of all authors are equal in selecting a data, calculating descriptors, analyzing results and writing a manuscript.
Competing interests
The authors declare that they have no competing interests.
Acknowledgment
The authors are thankful to Ho Chi Minh City University of Technology and Education for supporting websites to download the scientific articles.