

A computational study of cysteine and glutathione binding to small gold cluster Au8
- Computational Chemistry Research Group, Ton Duc Thang University, Ho Chi Minh City, Viet Nam
- Department of Chemistry, Can Tho University, Viet Nam
Abstract
Introduction: Understanding the binding mechanism between gold and is a fundamental step for numerous applications in biosensors and targeted drug delivery. This study aims to clarify the adsorption behaviors of CYS and GSH on the gold surface using a small gold Au8 cluster as a model reactant.
Methods: Here, we examine in details the molecular interaction between Au8 cluster with (CYS) and (GSH) by means of density functional theory (DFT). The PBE functional is employed in combination with the basis set for non-metal atoms and the basis set for gold. Harmonic frequencies are also computed to confirm optimized geometries as local minima or transition states on the potential energy surfaces.
Results: The calculated results show that these molecules prefer to anchor on the gold cluster via the sulfur atom with the adsorption energies of 20.3 and 30.8 / for CYS and GSH, respectively, in gas phase. In water, such values are considerably reduced, namely 19.0 / for CYS and 26.4 / for GSH. If a visible light with a frequency of v = 6x1014 Hz (500 nm) is applied, the time for the recovery of CYS and GSH from the most stable complexes will be about 1.24 and 6.03x107 seconds at 298 K in gas phase.
Conclusion: The Au8 cluster could be a promising material for designing sensor in CYS and GSH selective detection.
Introduction
In the past decades, gold clusters have been the subject of various studies owing to their potential applications in biosensors2, 1, catalysis3, biomedical dignostics5, 4, organic synthesis7, 6, environment4, drug delivery systems8, analysis9, electrochemistry11, 10. Recently, relevant studies have been carried out to elucidate the nature of interactions between biomolecules including amino acids, peptides, DNA constituents and small gold clusters, using both experimental and theoretical techniques.
According to Xie et al.12, the interaction between gold cluster Au (n = 3, 4) with cysteine and glycine is stabilized mainly due to Au-NH bond via the transfer of charge from the amine group to the Au atom. In another investigation on cysteine and larger gold Au cluster (n = 8, 10 and 12)13, the resulting complexes were found to be stabilized due to the strong Au−S bonding. Interactions of proline amino acid with Au cluster was also examined by DFT calculations14. Accordingly, two factors, i.e. the anchoring N-Au, O-Au bonds and the non-conventional O-HAu hydrogen bond, dominate the interactions. Other striking finding is that gold clusters prefer to anchor on the terminal amide group15, 14, 13.
In a study of M clusters (M = Ag, Au and Cu) binding to histidine in both neutral and anionic states, Javan et al.16 found that the metal clusters tend to oxidize histidine by getting electron from the lone pair orbitals of N, O, S atoms, and form highly stable anchor bonds. Scanning tunneling microscopy was also employed to probe the cysteine deposited on the Au(111) surface17. Novel network-like cluster structures of the layers with six and three cystine molecules on the Au surface have been found. A molecular dynamics simulation study of the adsorption mechanism of twenty amino acids and four surfactants on Au(111) in aqueous solution predicted an absorption energy around 3-26 kcal/mol18. The adsorption strength were found to correlate well with the degree of coordination of polarizable atoms (O, N, C) and determined by the molecular size/shape.
Cysteine and glutathione are among the most important thiol-containing compounds due to their special role in many biochemical reactions19. Cysteine (CYS) has many important cellular functions and its presence in biological fluids such as human plasma and urine is essential for clinical diagnostics of several diseases. The molecule on a solid surface is an important issue for either protein study or differentiating amino acids19. Previous study of cysteine adsorption on Au(100) showed the tendency of flat adsorption geometries due to the thiolate-amino bonds20.
Glutathione (GSH) or tripeptide γ-L-glutamyl-L-cysteinyl-glycine is well-known for its function in protecting the blood red cells from oxidative damage and maintaining the standard reduced state of the cells. In addition, the molecule is an integral part in the detoxification of the cells, responsible for elimination of harmful organic peroxides and free radicals. Accordingly, it absorbs toxins including pesticides, solvents, and heavy metals, then converting them into forms that are able to be excreted in urine or bile19. Moreover, its anionic form exhibits the particularly high tendencies to interact with gold clusters21. Recently, Thomas and co-workers22 introduced a novel approach for selective detection of cysteine and glutathione in many biological systems using gold nanorods.
Despite numerous efforts devoted to the absorption of such thiol-containing compounds on Au systems, the interaction mechanism is still not fully understood. Additionally, most of previous studies were performed in vacuum, while the effects of solvents have not been taken accounts. Understanding the interaction mechanism of gold nanoparticles with biomolecules is a fundamental step in designing and development of tiny biosensors.
This theoretical study aims to elucidate the adsorption behaviors of two thiol-containing cysteine and glutathione on the gold surface using small gold Au cluster as a model reactant. Current results could provide us with fundamental insights into the functionalization of gold nanoclusters with biomolecules and envisage their applicability in bio-chemical sensing and detection.
COMPUTIONAL METHODS
All calculations were performed with the aids of the Gaussian 09 program23. The geometries were fully optimized, making use of the functional PBE in conjunction with the basis sets cc-pVTZ-PP24 for gold and cc-pVTZ for non-metals.
Initial structures of the Au∙CYS and Au∙GSH complexes for geometry optimizations were created by attaching biomolecules via electron-rich centers, i.e. the S, N and O atoms, to the lowest-energy form of Au. Harmonic vibrational frequencies were also computed to verify that optimized geometries are true local minima and to estimate the zero-point vibrational energy (ZPE) corrections. Zero-point and thermal enthalpies corrections were then employed to predict free energies via the
where ΔE is the relative electronic energy at 0 K; ΔZPE is the relative vibrational energy at 0 K; ΔTCG is relative changes in Gibbs free energy going from 0 to 298 K. The complextion energy E of the interaction between Au cluster and CYS/GSH was computed as the value of energy difference:
where E is the lowest electronic energy of the X species. Hence, a negative value of E indicates a favorable adsorption. The greater the computed value of the complexation energy, the stronger is the affinity of cysteine binding to the gold cluster. The effect of solvent (water) was simulated using the default model IEF-PCM (Integral Equation Formalism-Polarizable Continuum Model)25 provided by Gaussian.
For deeper insights into the effect of interacting species on each other, the electronic properties such as the HOMO-LUMO energy gap (E) and the density of states (DOS) were examined. The E is a suitable aspect for defining the kinetic reactivity of materials26, and its conversion upon the adsorption process reflects the sensitivity of an adsorbent to an adsorbate. The GaussSum program27 has been used for DOS calculations.
RESULTS - DISCUSSION
Structural optimization
As expected, Au cluster prefers to interact with CYS or GSH species through electron-rich atoms, i.e. S, N and O atoms. These atoms with lone pair are more willing to form bonding with the 5d and 6s orbitals of the Au atom. At the PBE/cc-pVTZ/cc-PVTZ-PP level of theory, we located four conformations for Au×CYS complexes, denoted as C1-C4 (Figure 1), while for Au×GSH complexes, eight isomers G1-G8 (Figure 2) have been obtained. Noticeably, the geometries of both CYS, GSH and Au cluster are almost unmodified during the complexation. In addition, these structures have positive harmonic frequencies, indicating that they are local minima on the potential energy surfaces. The calculated complexation energies and bond lengths of the resulting complexes are listed in

Optimal structures located for the Au8∙CYScomplex in gas-phase (C1-C4). The relative values (kcal/mol) to the lowest-energy C1 conformation are given in brackets
Complexation energy (Ec, kcal/mol), Au-Y bond length (
| Conformation | Ec | Bond | |
| C1 | -20.27 | Au- | 2.395 |
| C2 | -18.50 | Au- | 2.520 |
| Au-O | 2.383 | ||
| C3 | -17.87 | Au- | 2.225 |
| Au-H | 2.156 | ||
| C4 | -14.06 | Au-O | 2.266 |
The most stable conformation of Au∙CYS complex, i.e. C1 form in Fig. 1, is formed by anchoring the thiol group on the low-coordinated gold atom of the Au ring. This can be understood by the hard-soft acid-base (HSAB) theory28, as the softer sulfur element is more willing to form strong bonds with soft elements like gold than the harder nitrogen and oxygen elements. In the C1 complex, the Au-S bond length is 2.395 Å, which is slightly longer than the sum of covalent radii of Au (1.35 Å) and S (1.02 Å) atoms. Previous study also predicted that Au atom exhibits a higher affinity with S atom29.
The second most stable species C2, which isconstructed by binding Auto the thiol and carbonyl groups of CYS is only about ~2 kcal/mol higher in energy than the C1 conformation. The Au-S and Au-O distances in C1 are 2.520 and 2.383 Å, respectively, which are quite larger than the sum of covalent radii of Au-S (2.370 Å) and Au-O (2.080 Å) bonds. The remaining conformations including C3 and C4 conformation are the most unstable conformations. In these conformations, they are formed by anchoring Au atom on CYS through N and O atoms, having stronger basicity than S atom. The second element is that their associated lengths are larger than the total of theirs covalent radii are 2.100 and 2.080 Å, respectively.

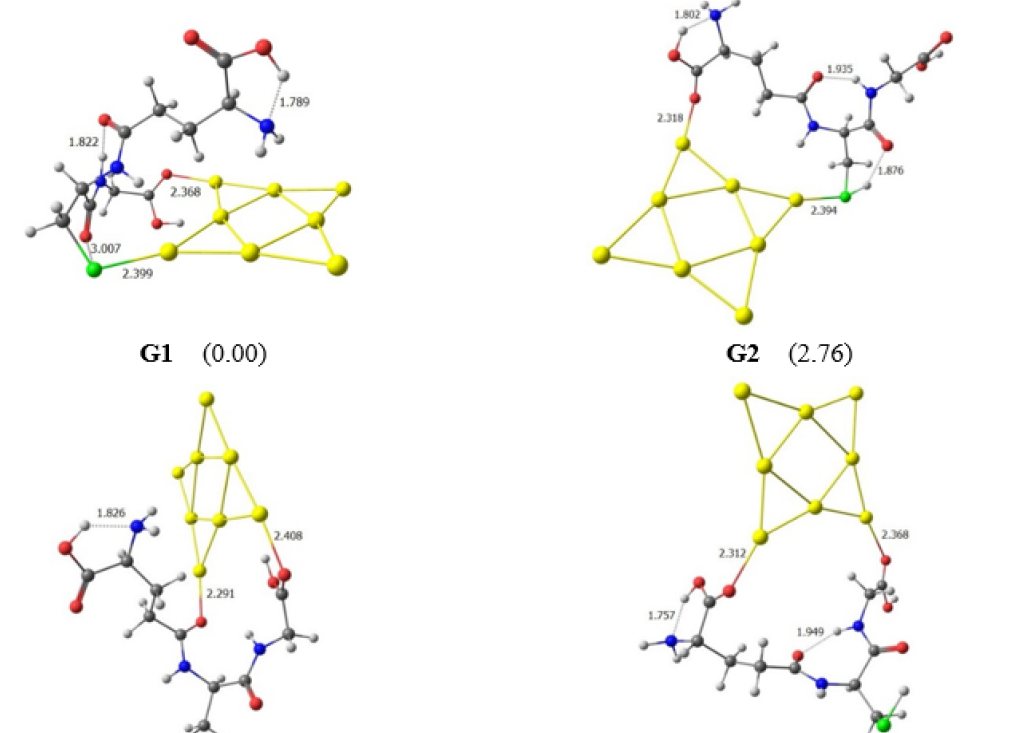
Optimal structures located for the Au8∙GSH complex in gas-phase (G1-G8). The relative values(kcal/mol) to the lowest-energy G1 conformation are given in brackets.
For the interaction between Au cluster and GSH molecules, eight conformations, i.e. G1-G8 in Figure 2, have been located. Among these, the most stable structure G1 is formed by anchoring the Au moiety on the S and O atoms of the thiol and carbonyl group. The Au atom has strong affinity with S atom, but the formation of inter-molecular hydrogen bonds (H-bonds) in GSH is also a vital factor that significantly contributes to stabilizing the Au.GSH complex21. In fact, in the G1 conformer, the calculated result obtain three H-bonds, namely N-HO=C, S-HO=C and COOHNH bonds. The H-bonds in G1 are much shorter than those of other products.
Complexation energy (Ec, kcal/mol), Au-Y bond length (
| Conformation | Ec | Bond | rAu-Y |
| G1 | -30.75 | Au- | 2.399 |
| Au-O | 2.368 | ||
| G2 | -27.99 | Au- | 2.394 |
| Au-O | 2.318 | ||
| G3 | -20.68 | Au-O | 2.291 |
| Au-O | 2.408 | ||
| G4 | -17.32 | Au-O | 2.312 |
| Au-O | 2.368 | ||
| G5 | -14.37 | Au-O | 2.491 |
| Au-O | 2.296 | ||
| G6 | -13.94 | Au-O | 2.328 |
| Au-O | 2.496 | ||
| G7 | -12.93 | Au-O | 2.545 |
| Au-O | 2.330 | ||
| G8 | -12.01 | Au-O | 2.264 |
| Au-H | 2.620 |
The next isomer G2 is only about ~3 kcal/mol less stable than the ground-state form G1. In G2, the Au atom links with cystenyl (Au-S) and glutamyl (Au-O), increasing the internal H-bonds in GSH. The remaining conformations (G3-G8) are quite unstable, being around 10-19 kcal/mol above the most stable form G1. In G3-G8 complexes, the gold cluster binding to GSH molecule through the O atoms of carbonyl groups.
Chemical reactivity
The quantum chemical descriptors of Aucluster, CYS and GSH molecules including chemical potential , total hardness and electrophile index are also examined to evaluate the chemical reactivity of systems considered. The calculated results at the PBE/cc-pVTZ/cc-pVTZ-PP level of theory are presented in
As expected, the Au has a more negative chemical potential than CYS and GSH molecules. In addition, the cluster exhibits a much larger electrophile index than the biomolecules. This strongly supports for the view that the metals are more willing to act as an electron-acceptor, while both CYS and GSH tend to be an electron-donor. Such a charge transfer from the biomolecules to the gold atoms results in a significant change in the HOMO-LUMO energy gap (E), which is reported as follows:
where and are the value of the for the bare Au cluster and the Au∙X (X = CYS, GSH) complexes, respectively. This parameter can also be used to evaluate the sensitivity of the clusters to the presence of biomolecules.
As shown in the plot of DOS (Figure 3), the band gap of Au in C1 (1.64 eV) and G1 (1.69 eV) considerably increase by 17% and 21%, respectively, as compared to that in the free Au (1.40 eV). In general, the HOMO energy of Au cluster is almost unchanged, while its LUMO energy substantially modified. In fact, due to an electron transfer from biomolecules to cluster, the LUMO of Au increased from -4.35 eV in free cluser to -3.92 eV and -3.84 eV in C1 and G1 complexes, respectively.
Quantum chemical descriptors of Au8 cluster, CYS and GSH at the PBE/cc-pVTZ/cc-pVTZ-PP level of theory
| Structures | EHOMO, eV | ELUMO, eV | μ , eV | η , eV | ω , eV | Eg, eV | ∆Eg, % |
| Au8 | -5.75 | -4.35 | -5.05 | 0.70 | 18.22 | 1.40 | - |
| CYS | -5.99 | -1.11 | -3.55 | 2.44 | 2.58 | 4.88 | - |
| GSH | -5.79 | -1.64 | -3.72 | 2.08 | 3.33 | 4.15 | - |
| C1 | -5.56 | -3.92 | -4.74 | 0.82 | 13.70 | 1.64 | 17.14 |
| G1 | -5.53 | -3.84 | -4.69 | 0.85 | 12.94 | 1.69 | 20.71 |

The DOS plot of C1 (top) và G1 (bottom) complexes.
Solvent effect and recovery time
Let us recall that all calculations above are performed in gas phase. In practice, sensors are often used to detect substances in solution, i.e. water environment. Therefore, further calculations have been carried out to examine the effects of water solvent on the interactions, using the IEF-PCM model (Integral Equation Formalism-Polarizable Continuum Model). The computed results are summarized in
In water, complexation energies, the changes of enthalpy and free energy become less negative, but also exhibit a similar trend as in gas phase. This is due to the fact that hydration energies of products are smaller than the sum of hydration energies of reactants. However, the interactions in water are still products favored with negative changes of free energies.
When exposed to the UV-Vis light, the biomolecules can undergo a desorption process. Very strong interactions are not favorable for chemical sensors because the desorption could be difficult and the sensor may suffer from long recovery times. According the transition state theory30, the recovery time (, seconds) exponentially depends on the binding energy Eas follows:
where T is the temperature, is the Boltzmann’s constant, is the attempt frequency and = 0.059 kcal/mol at 298 K.
Effect of water solvents on the C1 and G1 conformations
| Value | Au8 | C1 | G1 | |||
| Gas | Water | Gas | Water | Gas | Water | |
| Ec | - | - | -20.27 | -18.98 | -30.75 | -26.43 |
| ∆H | - | - | -19.15 | -17.20 | -30.77 | -25.94 |
| ∆G | - | - | -9.64 | -7.99 | -16.56 | -13.19 |
| HOMO | -5.75 | -5.21 | -5.56 | -5.18 | -5.53 | -5.16 |
| LUMO | -4.35 | -3.63 | -3.92 | -3.36 | -3.84 | -3.34 |
| Eg | 1.40 | 1.58 | 1.64 | 1.82 | 1.69 | 1.82 |
| % ∆Eg | - | - | 17.14 | 15.19 | 20.71 | 15.19 |
For the binding energy of -20.27 kcal/mol in vacuum, the recovery of cysteine molecules from the Au surface at 298 K is in the range of 0.025 to 1.7 seconds (
The time (in seconds) for the recovery of cys and gsh molecules from the Au8 surface at 298 K
| λ, nm | C1 | G1 | ||
| Gas | Water | Gas | Water | |
| 10 | 0.025 | 0.003 | 1.21x106 | 8.17x102 |
| 200 | 0.49 | 0.06 | 2.41x107 | 1.63x104 |
| 400 | 0.99 | 0.11 | 4.82x107 | 3.27x104 |
| 500 | 1.24 | 0.14 | 6.03x107 | 4.08x104 |
| 700 | 1.73 | 0.20 | 8.44x107 | 5.72x104 |
CONCLUSION
In this work, the structures, energetic properties and effects of water solution on the interaction of cysteine and glutathione molecules with the Au cluster was thoroughly examined by means of DFT calculations. The CYS and GSH moieties prefers to interact with the gold cluster through their thiol group. The nature of interactions is mainly determined by the charge transfer from the non-bonding pair of the sulfur atom to the empty orbitals of the gold cluster. Another noticable finding is that reactions of the gold cluster with these species are facile in nature, i.e. either molecular or dissociative chemisorptions are involved, rather than breaking molecular bonds and making again stronger bonds between the resulting fragments and the metals. The tendency to retain the initial forms of adsorbed molecules makes these clusters of great potential for applications in biochemical sensing and detection. If a visible light with a frequency of = 6 x 10 Hz (500 nm) is applied, the time for the recovery of CYS and GSH from the most stable complexes will be about 1.24 and 6.03 x 10 seconds at 298 K in gas phase. This indicates that Aucluster can be a promising candidate to develop tiny sensors in CYS and GSH selective detections.
COMPETING INTERESTS
The authors declare that there is no conflict of interest regarding the publication of this article.
AUTHORS’ CONTRIBUTIONS
All the authors contribute equally to the paper including the research idea and written manuscript.
ACKNOWLEDGMENT
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 103.01-2019.58. We are also grateful to the Interdisciplinary Center for Nano toxicity, Jackson State University, USA for using computing resources.