

Secretory expression of the recombinant FGF-2 protein in Pichia pastoris carrying multiple copies of target gene
- VNU-HCM University of Science
- VNUHCM – University of Science
Abstract
Introduction: Fibroblast growth factor-2 (FGF-2) is a multifunctional protein that plays an important role in the regulation of proliferation, differentiation and migration of a variety of cells. The recombinant human FGF-2 (rhFGF-2) is currently used in stem cell culture, medicine and cosmetic products. In this study, we aim to produce secreted rhFGF-2 protein from a Pichia pastoris strain containing multiple copies of the fgf-2 gene to eliminate the disadvantages of intracellular expression systems.
Methods: The recombinant Pichia pastoris carrying the fgf-2 gene was cloned by using homologous cloning method. The recombinant strains were screened by PCR reactions using specific primers for the target gene and the AOX1 promoter sequence. Moreover, the copy number of the fgf-2 gene inserted into the P. pastoris genome was identified by semi-quantitative PCR method. The secreted rhFGF-2 protein was collected in the induced BMMY medium at a final methanol concentration of 0.5%, and purified by one-step heparin affinity chromatography. The quantity and biological activity of the purified protein were determined by competitive ELISA method and MTT assay on NIH-3T3 cell line, respectively.
Results: Various recombinant P. pastoris clones carrying different copy numbers of the fgf-2 gene were obtained and categorized into 3 groups: the low copy strains (4-5 copies), medium copy strains (8-11 copies), and high copy strains (more than 20 copies). Among those strains, the 4-copy one produced the rhFGF-2 protein at the highest expression level. After purification, the purity of rhFGF-2 protein reached 98.8%, and the recovery yield was 179.2 µg of protein from 200 mL of flask culture (equivalent to 850 µg/L). The purified rhFGF-2 protein showed similar biological activity on NIH-3T3 cell line with the commercial FGF-2 protein.
Conclusion: The recombinant FGF-2 protein was successfully secretory expressed from Pichia pastoris, and successfully purified by only one-step chromatography.
Introduction
Basic fibroblast growth factor, also known as fibroblast growth factor-2 (FGF-2), was first discovered in 1973. It plays a crucial role in bone formation 1, regulation of tissue repair 2, 3, 4, wound healing 5, and angiogenesis 6. Currently, it is used in stem cell culture, medicine and cosmetics. To enhance the interaction between FGF-2 and its receptors, FGF-2 needs to bind to heparin or heparan sulfate proteoglycans 7, 8. The single chain of this protein contains 146 amino acids (pI = 9.6) and molecular weight is approximately 18 kDa. Specifically, it is easily expressed in prokaryote systems since the post-translational modifications and intra-disulfide bond are not required for its functionality. Up to now, FGF-2 has been expressed in various expression systems as intracellular protein 9, 10, 11, 12. However, these expression systems are not actually utilized since they have difficulty in disrupting the cells and purifying the target protein. The difficulties of intracellular expression systems have been mainly caused by taking time in collecting protein inside the cells and a lot of contaminating proteins of the host cells. Most of recombinant FGF-2 protein from intracellular expression systems need to perform 2-step purification in order to reach the expected purity of the desired protein, or tag cleavage step with fusion tag protein 9, 12. Hence, this study focused on using a secretory protein expression system of in order to overcome these disadvantages.
The methylotrophic yeast is the most common secretory heterologous recombinant protein expression system. This strain has the ability to secrete target proteins more efficiently than does,due to a strong promoter, tight regulation, and large size of exosomes 13, 14. Wild-type X33 strain possesses 2 genes, AOX1 and AOX2, which encode for the methanol metabolism protein. In particular, the AOX1 gene is regulated by the AOX1 promoter, which is a strong promoter. To enhance the yield of recombinant proteins in , the use of the AOX1 promoter is one of the most common strategies. There are two different types of homologous recombination-mediated insertion, which are (i) ends-in insertion which leads to additive insertion of the target gene and (ii) ends-out insertion which facilitates the replacement of the native AOX1 gene 15. The replacement of the AOX1 gene greatly reduces the ability of the host strain to consume methanol, and only one copy of the target gene is inserted into the genome, which can lead to low expression levels of the recombinant protein. Many studies have shown that the existence of multiple copies of recombinant genes can achieve better target protein expression levels 16, 17, 18. In addition, for this strain, it has been demonstrated that the expression level in optimized fermentation scale of the strain could be higher than shake-flask scale (approximately 16.4-fold) with simple and cheap culture medium 19. Thus, is a suitable host cell for industrial scale. Besides, the purification of secreted recombinant protein from might be easier than other systems since most of native secreted proteins have a pI lower than 6.0 20. In a previous study, FGF-2 was successfully expressed and purified in . The volumetric productivity of the purified FGF-2 (more than 94% of purity) reached 150 mg/L with optimized shake-flask culture condition and two-step purification; this result was 1.5 times higher than the yield of rhFGF-2 protein expressed in 9. However, this study did not investigate the effect of target gene copy on recombinant protein expression.
Therefore, in the present study, we constructed the recombinant X33 carrying multiple copy gene and further investigated the correlation between gene copy number and expression level of extracellular rhFGF-2 protein in . With the aim of shortening the downstream process and enhancing purity, the purification of the rhFGF-2 protein was experimentally performed with a 1-step purification strategy using heparin affinity chromatography. The biological activity of purified rhFGF-2 was evaluated on the NIH-3T3 cell line.
Materials-Methods
Gene, strains and plasmids
The gene encoding for FGF-2 was optimized from an origin sequence (UniProtKB - P09038) and then generated by overlap extension PCR method host strain DH5α (Thermofisher Scientific, California, US) and wild-type X33 (Thermofisher Scientific) were used for cloning and gene expression experiments, respectively. In addition, plasmid pPICZαA (Thermofisher Scientific) was used for cloning and expression studies.
Cloning of pPICZαA/plasmid in
The cDNA encoding for FGF-2 protein was amplified by PCR (Biorad, California, US) with specific primers FG-F and FG-R (primer sequences are shown in
Cloning of pPICZαA/plasmid in
In the present study, X33:: Mut wild type was used for cloning. The plasmid of positive colonies was extracted by alkaline-SDS lysis method, then transferred into X33 by electroporation (Biorad, California, US). Plasmid pPICZαA/ was integrated into host genome through recombinant homologous site on the AOX1 promoter sequence. To improve the recombinant homologous yield, the plasmid pPICZαA/ was linearized by I enzyme (Thermo Fisher Scientific). The transformants were selected on YPD-zeocin medium (Himedia) and further identified by PCR using the primer set of FG-F and FG-R. To investigate insertion type, the genome of recombinant was extracted and PCR was performed with the primer set of AOX-F and AOX-R.
Indirect quantification of recombinant fgf-2 gene copy number in
The genome of recombinant strains was extracted to perform PCR. The relative quantification of recombinant gene copy number was indirectly quantified by PCR products and compared with the standard curve. Wild-type X33:: Mut was used as a genome concentration standard. The number of gene copies was calculated by the ratio of the brightness of target gene band (Y) and AOX1 gene band (X) (Figure 1). The results of PCR were analyzed by electrophoresis and the brightness of band was determined by ImageJ software.

Illustration of indirect quantitative gene copy method by PCR and ImageJ. X, intensity of AOX1 gene band; Y, intensity of FGF-2 band.
Shake-flask expression in
A colony was picked up from the plate and overnight pre-cultured in 10 mL YPD-zeocin medium at 30C and 250 rpm. Then, the culture was transferred into 10 mL BMGY medium (Himedia) with a ratio of 1:10 (v/v). When OD reached 2.0 – 6.0, the cells were harvested by centrifugation at 5000 rpm (4C for 5 minutes), and the pellet collected. After that, the pellet was resuspended in 30 mL BMMY medium. In order to induce the expression of the gene, methanol (Xilong Scientific, Shantou, China) was added every 24 hours to a final concentration of 0.5% (v/v). After 72 hours, the supernatant of the culture was collected by centrifugation at 5000 rpm, 4C in 30 minutes. The rhFGF-2 expression was determined by SDS-PAGE gel (Himedia) using silver staining (Xilong Scientific) and competitive ELISA (Thermo Fisher Scientific).
Purification of rhFGF-2 by heparin-affinity chromatography
Considering the isoelectric point and specific affinity of the target protein, heparin-affinity chromatography was chosen to purify rhFGF-2. The HiTrap Heparin HP 5mL column (GE Healthcare, Chicago, US) was equilibrated with 25 mL Buffer A (20 mM Tris-HCl (Biobasic, Toronto, Canada), pH 7.5) at a rate of 5 mL/min. Subsequently, the supernatant was applied to the column at a rate of 5 mL/min. After that, the column was washed with 50 mL Buffer A. The rhFGF-2 protein was eluted by stepwise method with a variety of Buffer B (20mM Tris-HCl, pH 7.5, 2M NaCl (Scharlau, Barcelona, Spain)) concentration. The purity and yield of purification were determined by SDS-PAGE using silver staining, ELISA, and Gel Analyzer 2010a software.
Qualification of rhFGF-2 protein by SDS-PAGE and Western blotting
SDS-PAGE method was performed using a 15% gel according to the method of Laemmli. To detect the target protein expression, proteins in the gel were ran with low range protein marker (GE Health Care) and stained with 0.03% silver nitrate.

Construction of recombinant
Western blotting was performed to verify the presence of the rhFGF-2 protein. The in-gel protein after running SDS-PAGE was transferred onto nitrocellulose membrane (GE Health Care) and probed with mouse anti-human FGF-2 IgG (Sigma-Aldrich, Missouri, USA). The anti-mouse horseradish peroxidase (Sigma-Aldrich) was used as a secondary antibody. Detection of rhFGF-2 was carried out using Supersignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) and ImageQuant LAS 500 (GE Health Care).
Quantification of rhFGF-2 protein by competitive ELISA
The expression level of recombinant protein was measured by competitive ELISA method. After centrifugation, the supernatant of culture was collected. Before performing ELISA, the supernatant was diluted to lower protein concentration, depending on the protein concentration range of the standard curve, which was built with the commercialized rhFGF-2 (catalog number: 233-FB-025, R&D system, Minneapolis, US) as the standard. The supernatant was immobilized to the wells of a 96-well plate overnight. Unbound protein was washed 3 times with PBS-T and blanks on the well surface were filled with 2% BSA (Sigma-Aldrich) incubated for an hour. Mouse anti-human FGF-2 IgG (Sigma-Aldrich) and anti-mouse horseradish peroxidase (Sigma-Aldrich) were used as primary and secondary antibodies, respectively. Afterwards, an addition of TMB (Thermo Fisher Scientific) was performed to react with horseradish peroxidase. This reaction was stopped by HCl 2N (Scharlau, Barcelona, Spain). The concentration of rhFGF-2 was determined through the absorbance of the mixture at 450 nm by Multiskan Ascent (Thermo Fisher Scientific) and via the standard curve.
Analysis of biological activity of rhFGF-2
The bioactivity of FGF-2 was evaluated by its ability to stimulate the proliferation of NIH-3T3 cell line (ATCC), which has a number of FGF receptors on its cell surface. Based on NIH-3T3 cell numbers in culture medium, bioactivity of rhFGF-2 was compared with commercialized FGF-2 (catalog number: 233-FB-025, R&D system, Minneapolis, US) and negative control (without FGF-2 and without cell). The cell was cultured in DMEM/F12 without FBS (Sigma-Aldrich) and incubated with target protein for 36 hours. After this growth period, the cells were incubated with MTT solution (Sigma-Aldrich) for 4 hours to form insoluble formazan dye. After solubilization, the formazan dye was quantitated using Multiskan Ascent software. The measured absorbance directly correlates to the number of viable cells. This experiment was performed in triplicate, and the results were statistically analyzed by Graphpad Prism 6.0 software (GraphPad, Inc., CA, USA). The laboratory unit of proliferative bioactivity was calculated as follows:
Results
Construction of pPICZαA/plasmid
The gene encoding for FGF-2 was cloned into pPICZαA plasmid by eClone kit based on homologous recombinant mechanism. The recombinant vector was transferred into DH5α. The successfully transformed host cells were selected based upon their ability to grow on the LB medium containing zeocin, followed by colony PCR to determine the presence of the insert. The plasmid was extracted from the positive clones and further confirmed for the presence of the gene; the direction of the gene in the plasmid was confirmed by PCR with 2 different primer sets (Figure 2A). The PCR with the primer set of AOX5’-F and FG-R resulted in a single band of expected size for all 5 positive clones (Figure 2B). This implies that the gene was inserted after the AOX1 promoter and in the same orientation with the promoter. The PCR with the primer set of AOX5’-F and AOX3’-R was performed to make sure that only one copy number of gene was inserted (Figure 2B).
Cloning of pPICZαA/plasmid in
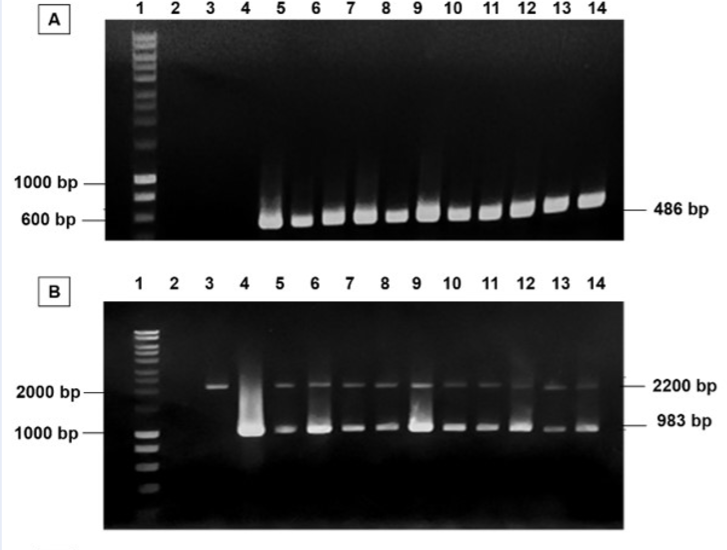
In this present study, the wild-type X33 was used for cloning. To enhance the homologous recombinant yield, pPICZαA/plasmidwas cut by I enzyme and then transferred into by electroporation. The positive transformants were selected through their ability to grow on YPD-zeocin medium and further subjected to colony PCR with primers in order to verify the presence of the pPICZαA/plasmid. The wildtype X33 and the pPICZαA/plasmid were used as negative and positive control, respectively. The PCR results showed that there was a single 486 bp band corresponding to a unique band of the positive control (Figure 3A). It indicated that carrying gene was successfully cloned.
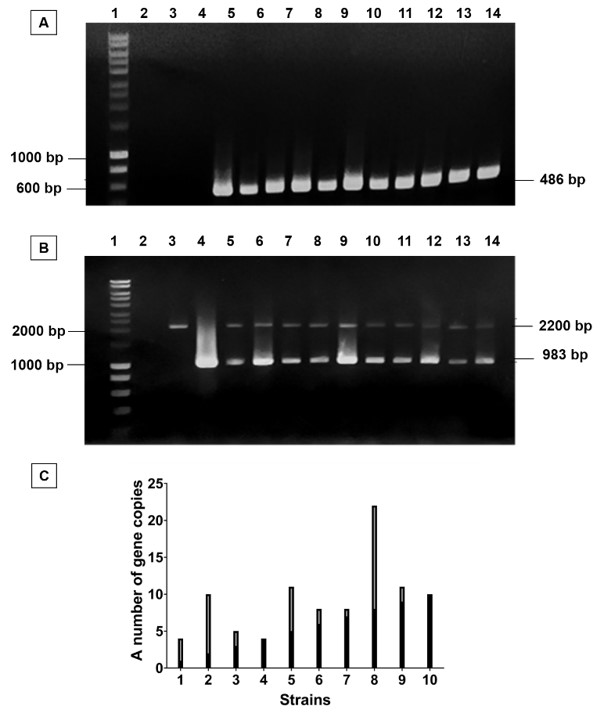
Cloning of pPICZαA/
Afterwards, the genotype of these transformants were determined by PCR with the primer set of AOX5’-F and AOX3’-R. The PCR results of all transformants showed that there were two bands at 2.2 kbp and 983 bp, equivalent to the length of AOX1 gene and gene with their own promoter, respectively. It revealed that all recombinant strains had additive insertion integration. Therefore, all strains were further determined for the gene copy number of each strain (Figure 3B). As the result of gene copy number quantification (Figure 3C), we succeeded in cloning multiple gene copy number of .
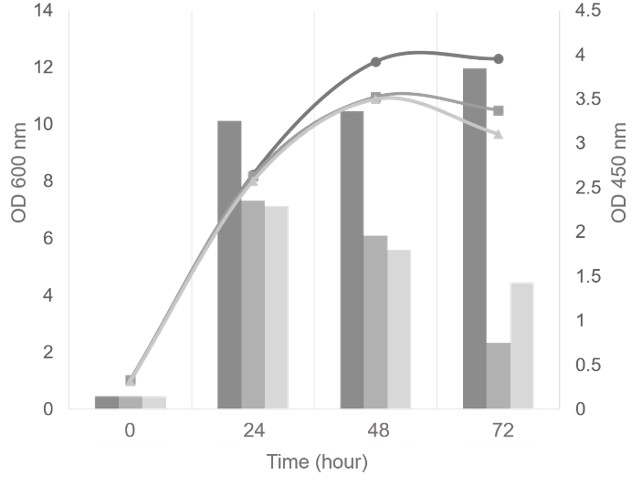
The growth and FGF-2 expression level of various
Comparison of protein expression among different gene copy strains
All strains were classified into three groups based on the copy number of gene: low copy strains (4-5 copies), medium copy strains (8-11 copies), and high copy strains (more than 20 copies). To investigate the effect of gene copy number variations on rhFGF-2 protein expression, one transformant of each group was cultivated in 100 mL BMMY medium and induced by 0.5% methanol. The gene copy number of each group was as follows: 4 (low), 11 (medium), and 22 (high) copies. Every 24 hours, the biomass was collected to measure OD and the supernatant was used to analyze rhFGF-2 expression by ELISA and SDS-PAGE. The results showed that all three strains had a common pattern of the growth curve (Figure 4). The highest OD reached 10-12 after 48 hours of cultivation. It indicated that the integrated gene copy number was not affected by the growth of this strain. Nevertheless, the expression levels among the 3 strains were considerably different (Figure 4). As the result, the expression level of the 4-copy strain increased slightly from 24 hours to the end of culture time. In contrast, the expression level of the 11-copy and the 22-copy strains dramatically decreased after 24 hours, despite the fact that growth was changed minimally. Moreover, the lowest copy strain reached a higher rhFGF-2 protein production than the others at 24, 48 and 72 hours, respectively. Consequently, this strain was selected to produce the rhFGF-2 protein, which later on would be studied further for the purification process and bioactivity assay.
Purification and identification of rhFGF-2
The X33:: was cultivated in 200 mL BMMY at 30C, 250 rpm and induced by 0.5% methanol. After 72 hours, the culture supernatant was collected in order to purify the rhFGF-2 protein by heparin affinity chromatography. The target protein was eluted by using a gradient from 0 to 100% of Buffer B. The protein purity and yield of purification were determined by SDS-PAGE and ELISA, respectively.
As seen from the SDS-PAGE results, there were two bands between 14.4 kDa and 20.1 kDa in the 60% - 90%B eluted fraction (well 4-6, Figure 5). These fractions were further confirmed by Western blotting by specific antibody (well 9-11, Figure 5) and revealed that both bands were the rhFGF-2 protein. The upper band was predicted to be the glycosylated rhFGF-2 protein.
The concentration of purified rhFGF-2 protein was qualified by ELISA based on the standard curve, and the purity was determined by Gel Analyzer software. The results showed that rhFGF-2 protein after purification reached 179.2 µg per 200 mL culture (equivalent to 850 µg/L) and the purity reached 98.8%. The recovery was estimated as approximately 6.13%.
In conclusion, the highly purified rhFGF-2 protein obtained from X33:: could accelerate the proliferation of mouse fibroblasts, equivalent to commercialized product, with laboratory units reaching 5.6 – 8.94 x 10 unit/ng.
Discussion
In this study, the low copy number of target gene resulted in the highest expression level which was similar to previous studies of multi-copy strains, even though the growth of the host strain was not affected by the integrated gene copy number. Many studies have demonstrated that multiple copy number of integrated gene could improve the expression level of the target protein. However, when the optimum is reached, further increase can sometimes cause negative effects 22, 23. For instance, high copy strain might suffer from protein folding-related oxidative stress and insufficient supply of carbon and energy sources 24.
As a result, the yield of FGF-2 protein in this study was lower than other expression systems in previous studies of 9, 21, where the culture condition was optimized for methanol concentration, pH, and induction time. Therefore, the efficiency of the strain in this study could be improved by optimized shake-flask condition 21 and be further enhanced in up-scale production, if dissolved oxygen, pH, temperature and carbon source feeding strategy could be controlled 19, 25. In short, might be a promising strain for high yield production of FGF-2 protein.

FGF-2 protein purification by heparin affinity chromatography using stepwise method. Lane 1: Protein ladder, Lane 2-6: SDS-PAGE result of eluted fraction at 50%, 60%, 70%, 80% and 90% of Buffer B, respectively, Lane 7-11: Western Blotting result of eluted fraction at 50%, 60%, 70%, 80% and 90% of Buffer B, respectively.
On the other side, the ease of purification in with specific affinity chromatography might bring more benefits than other intracellular expression systems 9, 26, due to lower content of extracellular proteins secreted from the host cells. As compared with another study of FGF-2 expressed from 21, the purification method of our study was better at reducing time and costs for downstream processes since we only performed one-step. The differences in results might be caused by the types of chromatography. Moreover, the recovery of purification in this study (6.13%) was higher than in a previous study with same type of chromatography (4.49%) 10. This might be affected by differences of equilibration and washing buffer. Briefly, heparin affinity chromatography is a rationale choice with good potential for shortening the purification process.
Bioactivity comparison of rhFGF-2 from
| Criteria | FGF-2 from P. pastoris | Commercialized FGF-2 | p-value |
| ED50 (ng/ml) | 14.52 ± 3.34 | 20.80 ± 3.60 | 0.6619 |
| Hill Slope | 1.202 ± 0.3196 | 1.689 ± 0.415 | 0.0566 |
| R2 | 0.9048 | 0.9577 | - |
| LU (unit/mg) | 5.6 – 8.94 x 104 | 4.10 – 5.81 x 104 | - |
After purification, two bands of FGF-2 protein were detected by Western blotting. The original FGF-2 protein is a non-glycoprotein; nevertheless, natural glycosylation processing can occur at hydroxy groups of threonine and/or serine residues in protein inside yeast cells 27. These amino acids were approximately 11.6% of the total peptide sequence of rhFGF-2 protein. Hence, we predicted that the upper band is glycosylated FGF-2 protein. The bioactivity of the mixture of glycosylated FGF-2 protein and non-glycosylated FGF-2 protein was demonstrated to be equivalent to commercialized FGF-2 protein, based on its ability to stimulate the proliferation of the NIH-3T3 cell line. Therefore, glycosylation did not affect the bioactivity of the FGF-2 protein.
Conclusions
In short, our study was successful in cloning multiple copy of gene into strain. In comparison with other multiple copy gene strains, the 4-copy gene strain has the ability to express the highest level of rhFGF-2 protein. The concentration of purified rhFGF-2 reached 179.2 µg per 200 mL of shake-flask culture (equivalent to 850 µg/L), with 98.8% purity after 1-step heparin affinity chromatography. In addition, the rhFGF-2 protein showed similar biological activity on the NIH/3T3 cell line as commercialized FGF-2 protein.
List of abbreviations used
AOX1: Aldehyde Oxidase 1
BMMY: Buffer Methanol-Complex Medium
bpP: Base pair
:
ELISA: Enzyme-linked immunosorbent assay
fgf-2: Fibroblast Growth Factor-2
kDa: Kilo Dalton
LB: Luria Broth
OD: Optical Density
PAGE: Polyacrylamide Gel Electrophoresis
:
PCR: Polymerase Chain Reaction
SDS: Sodium Dodecyl Sulfate
SDS-PAGE: Sodium Dodecyl Sulfate – Polyacrylamide Gel Electrophoresis
YPD: Yeast extract – Peptone – Dextrose medium
Acknowledgements
This work was supported by VNUHCM - University of Science research project funding, Laboratory of Molecular Biotechnology and Department of Molecular and Environmental Biotechnology infrastructure of VNUHCM - University of Science.
Author's contribution
This study was designed by Nguyen Hieu Nghia. Nguyen Cao Kieu Oanh contributed on data collection. Data analysis and interpretation for the work were carried out by Nguyen Hieu Nghia, Nguyen Cao Kieu Oanh and Le Kha Han. Nguyen Tri Nhan drafted the article and Le Kha Han wrote it. The article was critically revised and approved to be published by Nguyen Tri Nhan.
Conflicts of Interest
All author declare that they have no conflicts of interest.