

Triterpenoids and coumarins from the leaves of Sterculia foetida Linn.
- Faculty of Environmental Science, Sai Gon University, Ho Chi Minh City
- aculty of Environmental Science, Sai Gon University, Ho Chi Minh City
- Ca Mau Medical College, Ca Mau Province
- Duc Linh High School, Binh Thuan Province
- Chau Thanh High School, Ba Ria- Vung Tau Province
- Trang Bang High School, Tay Ninh Province
- epartment of Nature, Dong Nai University, Dong Nai Province
- Department of Chemistry, Ho Chi Minh City University of Education, Ho Chi Minh City
- Faculty of Chemistry, University of Science, Viet Nam National University Ho Chi Minh City, Ho Chi Minh City
- University of Medicine and Pharmacy at Ho Chi Minh City
Abstract
Introduction: Sterculia foetida Linn. is widely distributed in tropical countries. As the continuous study on the hexane and ethyl acetate extracts of Sterculia foetida leaves, the isolation and structural determination of four triterpenoids and two coumarins were addressed.
Method: The crude extract was prepared from dried power of Sterculia foetida leaves by maceration method in ethanol. This extract was then separated by liquid-liquid partition with n-hexane, chloroform, and ethyl acetate, respectively, to obtain the corresponding extracts. The hexane and ethyl acetate extracts were applied to multiple silica gel column chromatography to yield six compounds. Their chemical structures were determined by the NMR data analysis as well as the comparison their spectroscopic data and physical properties with those of reported literature.
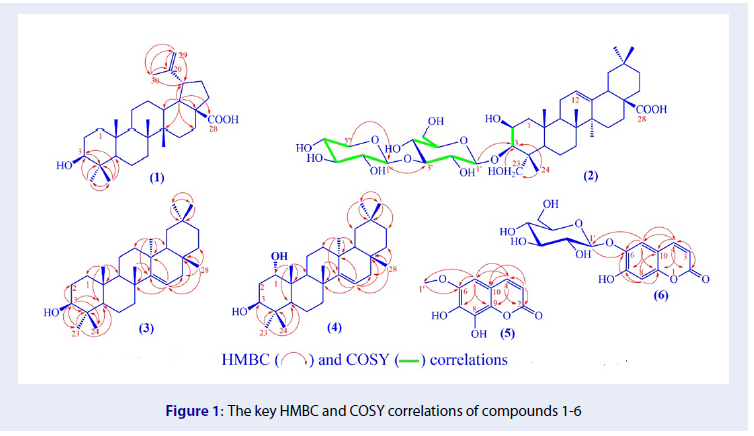
Results: Four triterpenoid compounds, including betulinic acid (1), conyzasaponin G (2), taraxerol (3), and taraxer-14-ene- 1a,3b -diol (4), and two coumarins fraxetin (5), and aesculin (6) were identified.
Conclusion: To the best of our knowledge, they have not been reported in the leaves of Sterculia foetida before, and compound 2 was known to present in Sterculia genus for the first time.