

Cytotoxic activities of synthesized curcumin and 3,4-Difluorinated curcumin against HepG2, LU-1 and KB cancer cell lines
- Faculty of Natural Sciences Pedagogy, Sai Gon University
- Faculty of Sciences, Nong Lam University
- Faculty of Chemical and Food Technology, Ho Chi Minh City University of Technology and Education
- Faculty of Environmental Science, Sai Gon University
Abstract
Introduction: Natural curcuminoids isolated from turmeric (Curcuma longa L.) have been limited in number and the amount of substrates evaluated in semi-synthetic processes and biological tests. Currently, potent anticancer activities of curcuminoids have garnered increased attention such that a greater number of synthetic procedures of curcumin analogues have been developed for further biological evaluations. The fluorine substituent of fluorinated compounds is important for biological responses. However, natural products bearing fluorine have rarely been found. In the study herein, we employed an aldol condensation between 4-hydroxy-3-methoxybenzaldehyde/3,4- difluorobenzaldehyde and pentane-2,4-dione to synthesize the desired curcumin (Cur) and 3,4- difluorinated curcumin (3,4-DFCur). Their half-maximal inhibitory concentration (IC50) values against HepG2, LU-1 and KB cancer cell lines were then assessed.
Methods: Pentane-2,4-dione was converted to enol form by using B2O3 before carrying out C-C coupling reactions with benzaldehyde analogues under basic conditions. The water scavenger was added to the reaction to capture the produced water. The reaction mixture was stirred at 70 ◦C. The reaction progress was monitored by thin layer chromatography (TLC). Crude products were purified by flash column chromatography (CC; SiO2, eluent: HEX/EA = 9/1!7/3). The chemical structures of the desired products were elucidated by 1H, 13C-NMR, HSQC and MS spectra. The anticancer activities of Cur and 3,4- DFCur against HepG2, LU-1 and KB cancer cell lines were determined using MTT method.
Results: Under reasonable reaction conditions, the yields for the coupling reactions were 53 and 72% for Cur and 3,4-DFCur, respectively. The stable enol tautomer of 1,3-diketone and the trans-configuration in a seven-carbon chain of product skeletons were assigned by 1H-NMR spectra. All synthesized products showed anticancer activities, with Cur exhibiting higher inhibitory activities when compared with 3,4-DFCur. Cur and 3,4-DFCur are Michael Acceptors; their cytotoxic activities could be attributed to the inhibition of glutathione S-transferase, a detoxification enzyme, by forming glutathionyl adducts. The decreased inhibitory capacities of 3,4-DFCur were due to the effect of fluorine which results in the unfavorable formation of reactive radicals and an increase in lipophilicity.
Conclusions: Curcumin and 3,4-difluorinated curcumin were completely synthesized in 53% and 72% yields. The synthetic procedure is applicable for synthesizing curcumin derivatives bearing various substituents on the aromatic rings, i.e., not limited to methoxy (-OCH3) and hydroxy (- OH) groups. Unexpectedly, the presence of fluorines in 3,4-DFCur led to lower cytotoxic activities against cancer cell lines. Our results provide greater insight on the structure-activity relationship of curcumin analogues against cancer cell lines.
INTRODUCTION
Cancer is a major public health problem worldwide and mortality rates from cancer have remained at the maximal peak1. There have been some success of chemotherapeutic drugs for treating cancer patients. However, the side effects from such medications have been reported numerous times in literature, and the impact from these medications can be severe and life-threatening. In order to explore drugs with more efficient properties yet fewer side effects, current research studies have focused on bioactive compounds found in plants as alternative treatments for cancer. Interestingly, curcumin (Cur), a ingredient isolated from turmeric (Curcuma longa L.), was studied as early as the 1940s. This polyphenol was shown to have anti-inflammatory, antioxidant and wound-healing effects.
Recent studies have shown that curcumin may be a potential anticancer agent2, 3, 4, 5, 6, 7, 8. Experimental findings on curcumin and pharmacophore modeling of its structure have revealed the substituents on phenol rings that are responsible for biological responses1, 9, 10, 11, 12. In order to explore novel curcumin analogues bearing various substituents, i.e., those not limited to methoxy (-OCH) and hydroxy (-OH) groups, a complete synthetic procedure of curcumin-related compounds was invented. Under basic condition, benzaldehyde derivatives were used as the starting materials to condense with pentane-2,4-dione to yield the desired curcuminoids5, 13, 14.
The effects of the fluorine substituent in molecules on the biological activities of those molecules are important and have been highlighted in previous literature15, 16. The responses can originate from alterations of the physicochemical properties, such as hydrogen bonding, and electronic properties of the fluorinated molecules.
In this study, we selected 4-hydroxy-3-methoxybenzaldehyde and 3,4-difluorobenzaldehyde as starting materials to generate C-C coupling reactions with pentane-2,4-dione in order to synthesize the desired curcumin and 3,4-difluorinated curcumin (3,4-DFCur) (Figure 1). Furthermore, in order to explore candidates (such as curcumin) with potential anticancer potencies, the cytotoxicity of those candidate agents were evaluated against human liver cancer (HepG2), lung cancer (LU-1), and human oral epidermal carcinoma (KB) cell lines. In these assays, the cytotoxicity of 3,4-DFCur was compared to that of Cur.

The synthetic procedure of curcumin (Cur) and 3,4-difluorinated curcumin (3,4-DFCur).
METHODS
Analytical methods
The chemical structures of Cur and 3,4-DFCur were elucidated by nuclear magnetic resonance (NMR) spectra, which were recorded on a Bruker Avance system (500 MHz (H), 125 MHz (C)). Mass spectrometry (MS) measurements were performed on an AGILENT 1200 series LC-MSD machine. The abbreviations of s (singlet), brs (broad singlet), d (doublet), dd (doublet of doublet), td (triplet of doublet), dt (doublet of triplet), t (triplet) and m (multiplet) were used to represent the patterns of the proton and carbon signals. The desired products were monitored and purified by using thin layer chromatograph (TLC; silica gel 60 F254; Merck) and flash column chromatography (CC; silica gel 0.035-0.070 mm; Merck). Sample spots on TLC were detected by UV light at λ = 254 and 366 nm. M5000 melting point meter (A. KRUSS, Germany) was used to measure the melting points of pure products with a heating rate of /min.
Synthetic procedure for Cur and 3,4-DFCur
Cur and 3,4-DFCur were synthesized according to a published procedure from literature13, 14. Pentane-2,4-dione (10.0 mmol) and boron oxide (10.0 mmol) were charged into a 100-mL two-neck round-bottom flask and stirred in ethyl acetate (20.0 mL) at for 45 min. The starting material- of 4-hydroxy-3-methoxybenzaldehyde (20.0 mmol) or 3,4-difluorobenzaldehyde (20.0 mmol)- and tri-n-butyl borate (40.0 mmol) were added together. The mixture reaction was stirred for 30 min to dissolve the aldehyde reactant before slowly adding n-butylamine (4.0 mmol) over 30 min into a solution via syringe. After completely adding the catalyst, the reaction was stirred at for 4-4.5 hrs. The reaction conversion was monitored by TLC. Finally, 20.0 mL of HCl solution (0.1 N) was added and stirred for 1 hr. The resulting mixture was extracted with DCM (3x40 mL). The combined organic phase was dried over NaSO and the solvent was removed under reduced pressure. The crude product was purified by flash CC (SiO, eluent: HEX/EA = 9/17/3).
Cytotoxic assay
Cur and 3,4-DFCur were tested in vitro for their cytotoxic activities against HepG2, LU-1 and KB cancer cell lines. The procedure was carried out by a modified MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich, St. Louis, MO, USA) method17, 18, 19. HepG2 (HB-8065), LU-1 (HTB-57), and KB (CCL-17) cell lines were obtained from ATCC (Manassas, VA, USA) and seeded in DMEM (Dulbeccos Modified Eagle Medium) (Sigma-Aldrich), supplemented with 10% FBS (Fetal Bovine Serum) (Sigma-Aldrich) and the other components (1% glutamine, 1% Penicillin and Streptomycin, Sigma-Aldrich). Cells were incubated in a humidified incubator (/ 5% CO). Ellipticine (0.01 M) was used as a positive control. All samples were completely dissolved in 10% dimethyl sulfoxide (DMSO) (Sigma-Aldrich) in medium. The appropriate concentration (190 L volume) of growing cells was added to each well of the 96-well plate. Sample solutions (10 L volume) were applied to culture wells to yield five final concentrations of 128, 32, 8, 2 and 0.5 g/mL in the wells. Each concentration was tested in three separate wells of a 96-well plate, and performed in triplicate. The cultures were incubated for 72 hours at 37 °C. Next, MTT (10 L, 5 mg/mL) was added to the wells. After 4 hours of incubation at 37 °C and subsequent removal of 210 L of culture medium from each well, the formazan product was dissolved in 100 L of DMSO. The optical density (OD) at 540 nm was measured with a BioTek microplate reader (Winooski, Vermont, USA).
The percent inhibition I (%) of inhibitory activity (see the Supporting Information for the I (%) values at various concentrations) was calculated using equation (1):
The IC (the half-maximal inhibitory concentration) was calculated by equation (2):
Where High and Low are high and low concentrations of samples, respectively. Highand Low refer to percent inhibition I (%) at High and Low.
In our assay, one compound was considered as inactive if its IC (g/mL) value was > 128 g/mL. Herein, the IC values of the tested compounds were presented in μM. TableCurve 2D was used as a statistical software to analyze data (Systat Software Inc).
RESULTS
Following published protocols, the desired Cur and 3,4-DFCur agents were synthesized in moderate yields by treatment of 4-hydroxy-3-methoxybenzaldehyde or 3,4-difluorobenzaldehyde with pentane-2,4-dione in the presence of boron oxide, n-butylamine, and tri-n-butyl borate in ethyl acetate at for 4-4.5 hours (
Isolated yields, reaction time and IC50 values (against HepG2, LU-1 and KB cancer cell lines) of Cur and 3,4-DFCur.
|
Compound |
Yield (%) |
Time (hr) |
IC50±SD[b] ( | ||
|
HepG2 |
LU-1 |
KB | |||
|
Cur |
53 |
4.5 |
35.47±2.77 |
72.88±6.19 |
33.35±2.66 |
|
3,4-DFCur |
72 |
4.0 |
42.69±3.56 |
305.04±24.50 |
66.36±5.80 |
|
Ellipticine[a] |
1.46±0.12 |
1.42±0.12 |
1.30±0.12 | ||
Cur: Yield 53% (1.95 g), red-orange solid, CHO [368.13 g/mol]; R = 0.31 (HEX/EA = 3/2); m.p. ; H-NMR (500 MHz, CDCl): δ = 3.96 (s, OCH, 3H), 3.95 (s, OCH, 3H), 6.42 (s, H, 1H), 6.83 (d, H, J (H,H) = 16.0 Hz, 1H); 6.93 (d, H, J (H,H) = 8.0 Hz, 1H), 6.94 (d, H, J (H,H) = 8.0 Hz, 1H), 6.99 (d, H, J (H,H) = 16.5 Hz, 1H), 7.02-7.08 (H, 4H), 7.11 (d, H, J (H,H) = 16.5 Hz, 1H), 7.29 (d, H, J (H,H) = 16.5 Hz, 1H) ppm. C-NMR (125 MHz, CDCl): δ = 55.9 (OCH), 55.9 (OCH), 97.6 (C), 108.2 (C), 108.8 (C), 110.9 (C), 113.8 (C), 114.6 (C), 114.8 (C), 121.5 (C), 121.6 (C), 128.2 (C), 128.5 (C), 134.8 (C), 135.6 (C), 146.7-146.9 (2C, 2C), 162.1 (C), 168.5 (C) ppm. ESI-MS m/z calc for [M+H]: 369.14; found: 368.90.
3,4-DFCur: Yield 72% (2.50 g), yellow solid, CHFO [348.08 g/mol]; R = 0.48 (HEX/EA = 95/5); m.p. ; H-NMR (500 MHz, CDCl): δ = 5.81 (s, H, 1H), 6.52 (d, H, J (H,H) = 16.0 Hz, 2H); 7.19 (m , H, 2H), 7.27 (m, H, 2H), 7.37 (m, H, 2H), 7.57 (d, H, J (H,H) = 16.0 Hz, 2H) ppm; C-NMR (125 MHz, CDCl):δ = 102.1 (C), 116.1-117.9 (C, C), 124.9 (C), 124.9 (C), 132.2 (C), 138.5 (C), 150.6 (d, C, J (C,F) = 247.5 Hz), 150.7 (d, C, J (C,F) = 248.7 Hz), 151.3 (d, C, J (C,F) = 251.2 Hz), 151.4 (d, C, J (C,F) = 251.2 Hz), 182.8 (C) ppm; ESI-MS m/z calc for [M+H]: 349.09; found: 348.80.
DISCUSSION
Chemistry
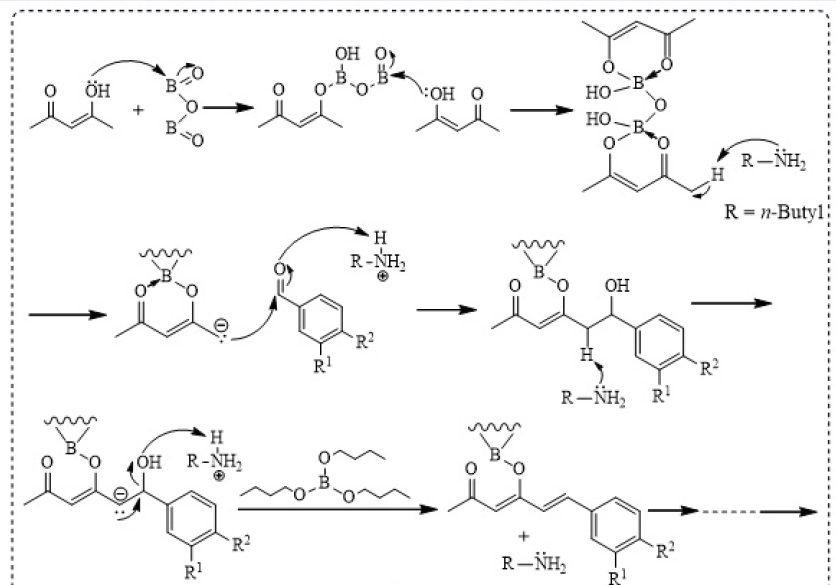
The reaction conditions- including solvent, catalyst, reaction time, complexing agent, water scavenger and temperature- play crucial roles on the synthetic yields of C-C coupling reactions between benzaldehyde analogues and pentane-2,4-dione, in the generation of Cur and 3,4-DFCur13, 14. The proposed mechanism of aldol condensation under basic condition was depicted in Figure 2. Firstly, in order to form the desired products, n-butylamine was selected as a basic catalyst to deprotonate the acidic methyl groups of pentane-2,4-dione. However, under basic condition, the 1,3-diketone moiety can undergo Knoevenagel condensation with aldehyde. Therefore, it was necessary to convert the 1,3-diketone form to an enolic structure. Boron oxide served as a complexing agent to form an enol configuration by complexing 1,3-diketone with boron. To minimize side reactions, the total amount of catalyst should be 40 mol % of 1,3-diketone, and the ratio of 2:1 for aldehyde and pentane-2,4-dione was employed. The formation of water in the reaction would reduce the yield. In order to avoid this, it was important to add a water scavenger to the reaction mixture.
Thus, tri-n-butyl borate was used as a water scavenger since it is an ester that would reduce water concentration via hydrolysis reaction. The reaction mixtures were stirred in ethyl acetate (as solvent) at 70 °C and, finally, the boron-Cur/3,4-DFCur complexes were cleaved by addition of a hydrochloric solution to generate the desired products.

Proposed mechanism for the synthetic procedures of curcumin (Cur) and 3,4-difluorinated curcumin (3,4-DFCur) using aldol condensation between benzaldehyde analogues and pentane-2,4-dione under basic catalyst.
The nature of substituents on the aromatic rings of benzaldehyde analogues affected the product yields (
The singlet signal at (ppm) 6.42 of Cur or 5.81 of 3,4-DFCur was assigned as a signal of proton bonded to the central carbon of a β-diketone. This observation indicated that the final products appeared as enol forms. In addition, two doublet signals were found in the range of 6.5 to 7.8 ppm, with coupling constants (J) of ~ 16 Hz, which characterize a resonance range of olefinic protons and pairs of trans-isomers. These results revealed trans-configuration in a seven-carbon chain of the curcumin skeleton.
In vitro cytotoxic activities
As summarized in
The anticancer activity of Cur correlated closely with the formation of reactive oxygen species (ROS)8, 22, 23, 24. The phenolic group of Cur can be abstracted H-atom to generate a phenoxyl radical which is stabilized by conjugation with adjacent electrons. The higher anticancer capacity of Cur may be attributed to the presence of para-substituted -OH group in the aromatic ring which results in the phenoxyl radical being involved in cell apoptosis. The replacement of fluorine atoms in the aromatic rings (3,4-DFCur) led to lower inhibitory activities compared with Cur. Our observations suggest that the fluorination caused the alterations on the electronic properties of curcumin structure. As mentioned, the phenolic motif is crucial for cell death. However, the formation of radicals is unfavorable for 3,4-DFCur. In addition, the absorption and penetration of inhibitors into the cell membrane are dependent on the hydrophobicity of the inhibitors. The fluorine substituent affects the physical properties of the inhibitor molecules, and aromatic fluorination increases their lipophilicity15. In the study herein, the results indicate that the lower activity of the fluorinated curcumin could be due to its increased lipophilicity.
CONCLUSION
Curcumin and 3,4-difluorinated curcumin have been completely synthesized in 53% and 72% yields from condensation reactions between benzaldehyde analogues and pentane-2,4-dione, using BO, tri-n-butyl borate and n-butylamine as complexing agent, water scavenger and basic catalyst, respectively. The moderate yields obtained from the synthetic procedures can be applied to synthesize curcumin and its derivatives since the isolation of natural curcuminoids from the Curcuma longa rhizome is a costly procedure and since organo-fluorine compounds isolated from plants have been virtually absent. The cytotoxicities against HepG2, LU-1 and KB cancer cell lines of the synthesized compounds were evaluated. The skeleton of the 4-hydroxy curcumin exhibited higher anticancer activities while aromatic substitutions by fluoro groups negatively impacted the anticancer activities. This was due to alterations of the electronic properties, which caused unfavorable formation of phenoxyl radicals, and the increased lipophilicity.
LIST OF ABBREVIATIONS
Cur: Curcumin
3,4-DFCur: 3,4-Difluorinated curcumin
DCM: Dichloromethane
EA: Ethyl acetate
HEX:n-Hexane
HepG2: Human liver cancer cell line
LU-1: Lung cancer cell line
KB: Human oral epidermal carcinoma cell line
IC: Half-maximal inhibitory concentration
NMR: Nuclear magnetic resonance
TLC: Thin layer chromatography
CC: Column chromatography
MS: Mass spectrometry
AUTHOR CONTRIBUTIONS
The contributions of all authors are equal in experimental design, laboratory work-up, data analysis and manuscript preparation.
COMPETING INTERESTS
The authors declare that they have no competing interests.
SUPPORTING INFORMATION
Supporting Information contains chemicals used for synthetic procedures and purifications of eight curcumin analogues, percent inhibition (I%) values at various concentrations and their NMR and MS spectra.
ACKNOWLEDGMENT
This work belongs to the project grant No: B2020-SPK-05 funded by Ministry of Education and Training, and hosted by Ho Chi Minh City University of Technology and Education, Vietnam.