

Alkaloids and phenolic compounds from the trunks of Coffea canephora and their alpha-glucosidase inhibition
- Department of Chemical Technology, Ho Chi Minh City University of Technology and Education, Ho Chi Minh City, Viet Nam
- Binh Loi Trung Secondary School, Ho Chi Minh City, Viet Nam
- Department of Chemistry, Ho Chi Minh City University of Education, Ho Chi Minh City, Viet Nam
Abstract
Introduction: Coffea canephora, known as Robusta coffee, is one of five coffee species grown in Vietnam. Continuous reporting of the chemical and bioactivity investigation of the Coffea canephora trunk, the isolation and spectroscopic analysis of alkaloids and phenolic glycosides, and their α-glucosidase inhibition were addressed.
Method: The crude methanol extract was prepared from dried Coffea canephora trunk powder using the maceration method. The n-hexane and ethyl acetate extracts were separated from the crude extract by sequence soaking in n-hexane and then in ethyl acetate. From these extracts, five organic compounds and a mixture were isolated using various chromatographic methods, and their structures were elucidated by NMR spectroscopic and MS techniques. αGlucosidase inhibition assays were carried out by Apostolidis’s method.
Results: Five compounds consisting of caffeine (1), uridine (2), 3,4,5-trimethoxyphenol (3), kelampayoside A (4), polystachyol (5), and a mixture of (±)-lyoniresinol 3 a -O-b-D-glucopyranoside (6) were isolated from the n-hexane and ethyl acetate extracts and structurally elucidated. αGlucosidase inhibition assays demonstrated that compounds 2, 4 and 5 showed better activity than acarbose.
Conclusion: To the best of our knowledge, all isolated compounds except compound 1 were investigated from the Coffea genus for the first time. Compounds 2, 4 and 5 showed better activity than acarbose in the α-glucosidase inhibitory assay.
INTRODUCTION
Coffea canephora Pierre ex A. Froehner, known as Robusta coffee, is one of five coffee species grown popularly in Vietnam1 and contributes significantly to the Vietnamese economy. Coffee beans and leaves have been investigated for chemistry and bioactivity aspects. Oestreich-Janzen’s study reported the presence of lipids, terpenoids, steroids, caffeic acid, chlorogenic acids, trigonelline, caffeine, and alkaloids in Coffea canephora.2 Metabolites isolated from coffee exhibit a reduced risk of Alzheimer’s3 and Parkinson’s diseases4 and anti-inflammatory5 and antioxidant activities6. In particular, the benefits of regular coffee consumption on the reduced risk of type 2 diabetes have been reported7. Previously, we reported the isolation, structural analysis and α-glucosidase inhibitory activity of triterpenoids and diterpenoids from the trunk of Coffea canephora.8, 9 In this article, two alkaloids and four phenolic glycosides were isolated from Coffea canephora trunk, and their α-glucosidase inhibitory ability was evaluated.
MATERIALS AND Methods
General experimental procedures
The NMR spectra were recorded on Bruker Avance at 500 MHz for H-NMR and 125 MHz for C-NMR, using residual solvent signal as internal reference (chloroform-d 7.260, 77.16, methanol-d 3.310, 77.16). The (HR)-ESI-MS was measured on a Bruker MicrOTOF-QII. The absorbance of p-nitrophenol at 405 nm for α-glucosidase inhibitory evaluation was obtained from a Biotex reader ELX800. Thin layer chromatography was carried out on precoated Kieselgel 60 F or silica gel 60 RP-18 F (Merck), and spots were visualized by spraying with 20% HSO solution, followed by heating. For column chromatography (CC), silica gel (Kieselgel 60, 0.040-0.063 mm, Merck), RP-18 (Merck), and Sephadex LH-20 (GE Healthcare) were used.
Plant material
Plant material, Coffea canephora Pierre ex A. Froehner was authenticated by Dr. Dang Van Son from the Institute of Tropical Biology, Vietnam Academy Science and Technology, Vietnam. A voucher specimen was deposited as UTE–A001 in the herbarium of Ho Chi Minh City University of Technology and Education, Vietnam. The Coffea canephora trunk for this research was collected in August 2018 in Lam Dong Province, Vietnam.
Extraction and isolation
The preparation of n-hexane and ethyl acetate extracts from Coffea canephora dried powdered trunk (30 kg) was described in our previous report. The n-hexane extract (H, 300 g) was chromatographed on a silica gel column using a gradient of an n-hexane:EtOAc mixture from 85:15 to 5:5 to afford seven fractions (H1–H7). Fraction H5 (3.55 g) was subjected to silica gel column chromatography (CC) three times, eluted with n-hexane:EtOAc (7:3), CHCl:MeOH (99:1) and n-hexane:acetone (8:2), purified further by Sephadex LH-20 CC, and eluted with CHCl:MeOH (1:4) to yield 3 (23 mg). The EtOAc extract (180 g) was subjected to silica gel CC eluted with n-hexane:EtOAc with increasing polarity from 50% to 100%, and then EtOAc:MeOH (9:1 to 8:2) was used to obtain 15 fractions (EA1-EA15). Fraction EA9 (20 g) was subjected to silica gel CC three times and eluted with EtOAc:MeOH (1:0, 95:5), EtOAc and CHCl:MeOH (98:2) to yield 1 (10 mg). Fraction EA10 (6.16 g) was fractionated using a gradient from 100% CHCl to 100% MeOH to afford five fractions (EA10.1-EA10.5). Fraction EA10.3 (0.276 g) was subjected to Sephadex LH-20 CC eluted with CHCl:MeOH (1:4) and then subjected to RP-silica gel CC using a mixture of MeOH:HO (4:6) as the eluent to yield 5 (6.1 mg). Fraction EA13 (8.31 g) was fractionated by Sephadex LH-20 CC eluted with CHCl:MeOH (1:4) to give 12 fractions (EA13.1–EA13.12). Fraction EA13.11 (0.693 g) was subjected to silica gel CC using an isocratic eluant of EtOAc:MeOH:HO (6:1:1) to furnish subfractions (EA13.11.1–EA13.11.5). Subfraction EA13.11.4 (259.7 mg) was subjected to RP-18 reversed-phase CC (MeOH:HO, 3:7) and then normal-phase silica gel CC (CHCl:MeOH:HO 20:6:1) and was purified further by Sephadex LH-20 CC to obtain compound 6 (5.4 mg). The same procedure was applied to subfraction EA13.11.5 (22.4 mg) to yield 4 (4.6 mg). Fraction EA14 (8.19 g) was fractionated by silica gel CC using CHCl:MeOH gradient elution (1:0 to 0:1), subjected to a Sephadex LH-20 column eluting with CHCl:MeOH (1:4), and further separated by silica gel CC eluted with CHCl:MeOH:HO (20:6:1) to yield 2 (4.2 mg).
α-Glucosidase assay
The α-glucosidase inhibitory activity assay was applied to compounds 1, 2, 4, 5, and 6 (triplicated) and carried out as described previously by Apostolidis10 with some modifications described in our previous report.
Dimethyl sulfoxide was used to dissolve the samples at different concentrations before being added to a 96-well plate. Each well contained 20 µL of the sample solution, 60 µL of the phosphate buffer solution (0.1 M, pH 6.8), 100 µL of the 200 µM p-nitrophenyl-D-glucopyranoside, and 20 µL of α-glucosidase (0.3 IU/mL). The reaction was conducted at 37°C for 30 minutes. The reaction was ended by adding 50 µL of 50 mM NaOH to each well. Acarbose was used to replace the sample solution for the positive control. Using a BioTek microplate reader, the absorbance of the samples was measured at 405 nm. Based on equation (1), the percentage of α-glucosidase inhibition, I (%), was calculated. The calibration curve equation of I (%) according to sample concentration was used to calculate the IC (the half-maximal inhibitory concentration). The differences in the mean values were analyzed using the Duncan test (p<0.05).
where A and A are the absorbances of the control and sample, respectively.
RESULTS
From the n-hexane and ethyl acetate extracts prepared from the trunk of Coffea canephora grown in Lam Dong Province, Vietnam, six compounds, including 1 (10 mg), 2 (4.2 mg), 3 (23 mg), 4 (4.6 mg), 5 (6.1 mg), and 6 (5.4 mg), were isolated by chromatographic methods. The spectroscopic data of compounds 1-4 are displayed as follow, while the data of compounds 5 and 6 are shown in
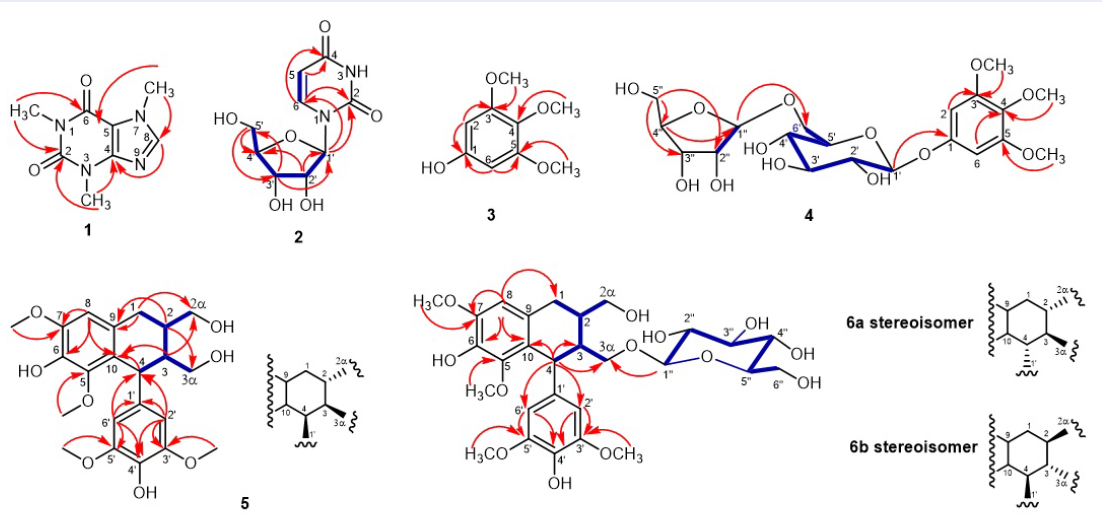
Caffeine(1): White amorphous powder. ESI-MS: m/z 194.8 [M+H] (calcd. for CHNO+H, 195.1). H-NMR data (CDCl, 500 MHz) (δ, J in Hertz): 7.51 (1H, s, H-8), 4.00 (3H, s, 7-CH), 3.59 (3H, s, 3-CH), 3.41 (3H, s, 1-CH). C NMR data (CDCl, 125 MHz): δ 155.4 (C-6), 151.7 (C-2), 148.7 (C-4), 141.4 (C-8), 107.6 (C-5), 33.6 (7-CH), 29.7 (3-CH), 27.9 (1-CH). Selected HMBC correlations are depicted in Figure 1.
Uridine(2): Yellowish amorphous powder. ESI-MS: m/z 244.7 [M+H] (calcd. for CHNO+H, 245.1). H-NMR data (CDOD, 500 MHz) (δ, J in Hertz): 7.99 (1H, d, 8.0, H-6), 5.89 (1H, d, 4.5, H-1'), 5.69 (1H, d, 8.0, H-5), 4.17 (1H, dd, 10.0, 5.5, H-2'), 4.15 (1H, dd, 10.0, 5.0, H-3'), 4.00 (1H, m, H-4'), 3.84 (1H, dd, 12.0, 2.5, H-5'a), 3.73 (1H, dd, 12.0, 3.0, H-5' ). C NMR data (CDOD, 125 MHz): δ 166.2 (C-4), 152.5 (C-2), 142.7 (C-6), 102.7 (C-5), 90.8 (C-1'), 86.4 (C-4'), 75.7 (C-2'), 71.3 (C-3'), 62.3 (C-5'). Selected HMBC and COSY correlations are depicted in Figure 1.
3,4,5-Trimethoxyphenol (3): Yellowish amorphous powder. HR-ESI-MS: m/z 207.0630 [M+Na] (calcd. for CHO+Na, 207.0633). H-NMR data (CDCl, 500 MHz) (δ, J in Hertz): 6.08 (2H, s, H-2/6), 3.78 (3H, s, 4-OCH), 3.77 (6H, s, 3/5-OCH). C-NMR data (CDCl, 125 MHz): δ 153.9 (C-3/5), 152.7 (C-1), 131.9 (C-4),93.2 (C-2/6), 61.2 (4-OCH), 56.1 (3/5-OCH). Selected HMBC correlations are depicted in Figure 1.
Kelampayoside A (4): White amorphous powder, HR-ESI-MS: m/z 501.1563 [M+Na] (calcd. for CHONa, 501.1579). H NMR data (CDOD, 500 MHz) (δ, J in Hertz): 6.46 (2H, s, H-2/6), 4.97 (1H, d, 2.5, H-1″), 4.79 (1H, overlapped, H-1'), 4.04 (1H, dd, 11.0, 2.0, H-6'a), 3.95 (1H, d, 10.0, H-4″a), 3.88 (1H, d, 2.5, H-2″), 3.82 (6H, s, H-3/5), 3.74 (1H, d, 9.5, H-4″b), 3.71 (3H, s, 4-OCH), 3.61 (1H, m, H-6'b), 3.56 (1H, m, H-5'), 3.55 (2H, s, H-5″), 3.44 (1H, m, H-3'), 3.42 (1H, m, H-2'), 3.32 (1H, H-4' C NMR data (CDOD, 125 MHz): δ 155.9 (C-1), 154.8 (C-3/5), 134.8 (C-4), 110.9 (C-1″), 103.2 (C-1'), 96.5 (C-2/6), 80.5 (C-3″), 78.0 (C-2″), 77.9 (C-3'), 77.0 (C-5'), 75.0 (C-2'), 74.9 (C-4″), 71.6 (C-4'), 68.7 (C-6'), 65.5 (C-5″), 61.3 (4-OCH), 56.8 (3/5-OCH). Selected HMBC and COSY correlations are depicted in Figure 1.
Polystachyol (5): Yellowish gum. HR-ESI-MS: m/z 443.1671 [M+Na] (calcd. for CHONa, 443.1676). H-NMR data (CDOD, 500 MHz) and C-NMR data (CDOD, 125 MHz) are presented in
(±)-Lyoniresinol 3α-O--D-glucopyranoside (6): Yellowish gum. HR-ESI-MS: m/z 605.2216 [M+Na] (calcd. for CHONa, 605.2205). H-NMR data (CDOD, 500 MHz) and C-NMR data (CDOD, 125 MHz) are presented in
|
Compounds |
IC50 ( |
|
1 |
393.60 ± 0.10 |
|
2 |
202.40 ± 0.04 |
|
4 |
182.87 ± 0.02 |
|
5 |
139.51 ± 0.02 |
|
6 |
N/A |
|
Acarbose |
209.80 ± 0.01 |
NMR data of compound 5 and mixture 6
|
Position |
Compound 5 (CD3OD) |
6a stereoisormer (CD3OD) |
6b stereoisormer (CD3OD) | |||
|
δH, mult. (J, Hz) |
δC |
δH, mult. (J, Hz) |
δC |
δH, mult. (J, Hz) |
δC | |
|
1 |
2.70 dd (15.0, 4.5) 2.57 dd (15.0, 11.5) |
33.6 |
2.72 dd (15.0, 4.5) 2.61 dd (15.0, 11.5) |
33.8 |
2.67 m |
33.8 |
|
2 |
1.63 m |
40.9 |
1.70 m |
40.7 |
1.70 m |
41.3 |
|
2α |
3.59 dd (11.0, 5.0) 3.48 dd (10.5, 7.0) |
66.8 |
3.63 m 3.54 dd (11.0, 6.5) |
66.3 |
3.63 m 3.54 dd (11.0, 6.5) |
66.3 |
|
3 |
1.97 m |
49.5 |
2.10 m |
46.7 |
2.14 m |
46.6 |
|
3α |
3.50 d (5.5) |
64.2 |
3.88 m 3.46 dd (10.0, 4.0) |
71.6 |
3.88 m 3.60 m |
72.1 |
|
4 |
4.31 d (5.5) |
42.3 |
4.42 d (6.0) |
42.8 |
4.23 d (6.5) |
43.2 |
|
5 |
- |
147.7 |
- |
147.6 |
- |
147.6 |
|
6 |
- |
138.9 |
- |
138.9 |
- |
138.9 |
|
7 |
- |
148.7 |
- |
148.7 |
- |
148.7 |
|
8 |
6.59 s |
107.8 |
6.58 s |
108.0 |
6.58 s |
107.9 |
|
9 |
- |
130.2 |
- |
130.3 |
- |
130.3 |
|
10 |
- |
126.3 |
- |
126.4 |
- |
126.3 |
|
1' |
- |
139.3 |
- |
139.3 |
- |
139.4 |
|
2'/6' |
6.38 s |
107.0 |
6.43 s |
107.1 |
6.41 s |
107.3 |
|
3'/5' |
- |
149.0 |
- |
149.0 |
- |
149.0 |
|
4’ |
- |
134.6 |
- |
134.6 |
- |
134.7 |
|
5-OCH3 |
3.38 |
60.2 |
3.35 s |
60.2 |
3.33 s |
60.2 |
|
7-OCH3 |
3.86 s |
56.6 |
3.86 s |
56.7 |
3.86 s |
56.7 |
|
3'/5'-OCH3 |
3.74 s |
56.8 |
3.75 s |
56.9 |
3.75 s |
57.0 |
|
1'' |
- |
- |
4.29 d (8.0) |
104.8 |
4.14 d (8.0) |
104.3 |
|
2'' |
- |
- |
3.22 m |
75.2 |
3.22 m |
75.1 |
|
3'' |
- |
- |
3.37 m |
78.3 |
3.31 m |
78.2 |
|
4'' |
- |
- |
3.31 m |
71.7 |
3.31 m |
71.7 |
|
5'' |
- |
- |
3.25 m |
78.0 |
3.16 m |
78.0 |
|
6″ |
- |
- |
3.65 dd (12.0, 5.5) 3.83 m |
62.9 |
3.69 dd (12.0, 5.5) 3.83 m |
62.8 |
DISCUSSION

Selected HMBC correlations of compounds 1-6
Compound 1isolated from fraction EA9 was a white amorphous powder. The H-NMR spectrum of compound 1 exhibited a singlet signal at δ 7.51 (1H, s, H-8) for a characteristic aromatic proton on a fully substituted aromatic system. Three singlet signals at δ 3.41 (3H, s, 1-CH), 3.59 (3H, s, 3-CH), and 4.00 (3H, s, 7-CH) revealed the presence of three methyl groups attached to heteroatoms. Its C-NMR displayed 8 carbons, including two carbonyl carbons resonated at δ 155.4 (C-6) and 151.7 (C-2), three olefinic carbons atδ 107.6 (C-5), 148.7 (C-4), and 141.4 (C-8), and three methyl groups at δ 27.9 (1-CH), 29.7 (3-CH), and 33.6 (7-CH). The skeleton of compound 1 and the positions of the three methyl groups were determined by the HMBC correlations displayed in Figure 1. Its ESI-MS showed the molecular ion peak [M+H]at m/z 194.8 (calcd. for CHNO+H, 195.1) corresponding to the molecular weight of caffeine, an abundant compound in coffee beans. The NMR data of compound 1 were compatible with those in Sitkowski’s report11. Therefore, compound 1 was identified as caffeine.
Compound 2 was isolated as a yellowish amorphous powder from fraction EA14. Downfield, the H-NMR spectrum of 2 displayed two olefinic methine signals coupling each other at δ 7.99 (d, 8.0 Hz, H-6) and δ 5.69 (d, 8.0 Hz, H-5). Four oxymethine signals resonated at δ 5.89 (d, 4.5 Hz, H-1'), 4.17 (dd, 10.0, 5.5 Hz, H-2'), 4.15 (dd, 10.0, 5.0 Hz, H-3') and 4.00 (m, H-4') along with two oxymethylene signals at δ 3.84 (dd, 12.0, 2.5 Hz, H-5'a) and 3.73 (dd, 12.0, 3.0 Hz, H-5'b) were features of a pentose. The C-NMR spectrum of compound 2 showed nine carbons, including two carbonyl groups at δ 166.2 (C-4) and 152.5 (C-2), two olefinic methines at δ 142.7 (C-6) and 102.7 (C-5), four oxymethines at δ 90.8 (C-1'), 86.4 (C-4'), 75.7 (C-2') and 71.3 (C-3'), and an oxymethylene at δ 62.3 (C-5'). The signals at δ 5.89 (d, 4.5 Hz, H-1') and δ 90.8 (C-1'), which were characteristic of an anomer position, indicated the presence of a sugar unit. This fact was supported by the COSY correlations of H-1'–H-2'–H-3'–H-4'–H-5'. The HMBC cross-peaks displayed in Figure 1 allowed us to determine a ribose unit for compound 2. Two olefinic methines belonging to the same ring with two carbonyl groups were demonstrated using COSY correlations between δ 7.99 (H-6) and 5.69 (H-5) and HMBC correlations from H-6 (δ 7.99) to C-2 (δ 152.5) and C-4 (δ 166.2) and from H-5 (δ 5.69) to C-4 (δ 166.2). In addition, based on the HMBC cross-peaks from H-1' (δ 5.89) to C-6 (δ 142.7) and C-2 (δ 152.5), the ribose was determined to attach the aglycon at C-1'. The H-NMR and C-NMR data of compound 2 were compatible with those of uridine12. The ESI–MS displayed the pseudomolecular ion peak at m/z 244.7 [M+H] which was appropriate for uridine (calcd. for CHNO+H, 245.1). The above information supported the identification of compound 2 as uridine.
Compound 3 was isolated from fraction H5 as a yellowish amorphous powder. Its HR-ESI-MS showed a sodiated molecular peak at m/z 207.0630 [M+Na] appropriate for a calculated molecular weight of CHONa of 207.0633. Therefore, the molecular formula of compound 3 was assigned to CHO. Its H-NMR spectrum exhibited a singlet at δ 6.08 (s, H-2/6) for aromatic protons. In addition, two singlets resonated at δ 3.78 (s, 4-OCH) and 3.77 (s, 3/5-OCH), suggesting that the aromatic ring bears three methoxy groups. The C-NMR spectrum of 3 displayed seven signals, including 2 aromatic methines at δ 93.2 (C-2/6), 4 oxygenated aromatic carbons at δ 153.9 (C-3/5), 152.7 (C-1), 131.9 (C-4), and 3 methoxy groups at δ 61.2 (4-OCH), 56.1 (3/5-OCH).The locations of methoxy and hydroxyl groups were determined by HMBC correlations, which are displayed in Figure 1. The 1D-NMR data of compound 3 had good compatibility with 3,4,5-trimethoxyphenol13. Therefore, the structure of compound 3 was elucidated as 3,4,5-trimethoxyphenol.
Compound 4 was isolated as a white amorphous powder from fraction EA13. Its molecular formula was identified as CHO using HR-ESI-MS with a quasi-molecular ion peak [M+Na] at m/z 501.1563 (calc. for CHO+Na, 501.1579). The H-NMR and C-NMR spectra of compound 4 were similar to those of compound 3 in the presence of signals indicating a tetrasubstituted aromatic ring, except for the addition of two sugar moieties. The presence of two sugar units was demonstrated by two anomer protons at δ 4.79 (H-1') and 4.97 (d, 2.5 Hz, H-1″) and oxygenated protons in the zone of 3.0-4.0 ppm. The signal at δ 4.79 (H-1') overlapping with the residual water signal was assigned using the HSQC spectrum. Additionally, a quaternary carbon at δ 134.8 (C-4) was explored by the HMBC spectrum. The analysis of the C-NMR spectrum aligned with the H-NMR and HSQC spectra along with the COSY spectrum allowed the assignment of six oxygenated carbons at δ 103.2 (C-1'), 75.0 (C-2'), 77.9 (C-3'), 71.6 (C-4'), 77.0 (C-5'), 68.7 (C-6') belonging to a hexose moiety and five remaining oxygenated carbons at δ 110.9 (C-1″),78.0 (C-2″), 80.5 (C-3″), 74.9 (C-4″) and δ 65.5 (C-5″) belonging to a pentose unit. The attachment of two sugar units to the aromatic ring was assigned by the HMBC correlations (Figure 1). From the above analysis, compound 4 was predicted to be a phenolic diglycoside. Based on a previous report14, the NMR data of compound 4 showed good compatibility with 3,4,5-trimethoxyphenyl 1-O-β-D-apiofuranosyl(1→6)-glucopyranoside or kelampayoside A and was elucidated as this structure.
Compound 5 was isolated as a yellowish gum from fraction EA10. The HR-ESI-MS spectrum exhibited a sodiated molecular ion peak [M+Na]at m/z 443.1671 (calcd. for CHO+Na, 443.1676) to identify the molecular formula of compound 5 as CHO with 9 degrees of unsaturation. Its H-NMR displayed two singlet signals at δ 6.59 (s, H-8) and δ 6.38 (s, H-2'/6'), indicating the presence of a 1,2,3,4,5-pentasubstituted ring and a 1,3,4,5-tetrasubstituted ring. In addition, three singlet signals at δ 3.38 (s, 5–OCH), 3.74 (s, 3'/5'–OCH) and 3.86 (s, 7–OCH) were assigned to four methoxy groups. The signals at δ 3.50 (d, 5.5 Hz, H-3α), 3.59 (dd, 11.0, 5.0 Hz; H-2αa) and 3.48 (dd, 10.5, 7.0 Hz; H-2αb) demonstrated the presence of two oxymethylene . The signals at δ 2.70 (dd, 15.0, 4.5 Hz, H-1a), 2.57 (dd, 15.0, 11.5 Hz, H-1b), 1.63 (m, H-2), 4.31 (d, 5.5 Hz, H-4) and 1.97 (m, H-3) were characteristic of an aliphatic fragment. The C-NMR along with H-NMR and HSQC indicated that compound 5 had 22 carbons, including two oxymethylenes at δ 64.2 (C-3α) and 66.8 (C-2α), four methoxy groups at δ 60.2 (5–OCH), 56.6 (7–OCH), and δ 56.8 (3'/5'–OCH), a methylene at δ 33.6 (C-1), three methine at δ 49.5 (C-3), 42.3 (C-4), and 40.9 (C-2), and 12 aromatic carbons. The COSY spectrum supported the identification of two associated proton spin systems of H-1–H-2–H-2α and H-4–H-3–H-3α connecting each other at C-2–C-3, which are depicted in Figure 1. From the above information, compound 5 was predicted to have two benzene rings connected via a side chain with the feature of a tetrahydronaphthalene lignan or aryl-tetralin-type lignan. The HMBC cross-peaks from the aromatic protons and the methoxy protons to the aromatic carbons supported the identification of two aromatic ring systems as 4-hydroxy-3,5-dimethoxyphenyl moieties. The attachment of two aromatic rings to the side chain was established by HMBC correlations, which are displayed in Figure 1. The HMBC correlations from H-1 and H-2 to C-9, from H-3 to C-10, and from H2'/6' to C-4 indicated the connection between the two rings to the side chain. The combination of the above NMR analysis and comparison of NMR data with a previous report15 demonstrated the structure of compound 5 as polystachyol.
Mixture 6 was isolated from fraction EA13. The molecular formula of 6 was established as CHO using HR-ESI-MS with a sodiated molecular ion peak [M+Na] at m/z 605.2216 (calcd. as 605.2205). The H-NMR and C-NMR spectra of 6 were similar to those of compound 5 except for an additional set of glucopyranosyl signals in the zone 3.0-4.0 ppm according to 62-105 ppm, along with anomeric protons at 4.29 (d, 8.0 Hz, H-1″, 6a) and 4.14 (d, 8.0 Hz; H-1″, 6) and two acetal carbons at 104.8 and 104.3. Using the HMBC experiment, the location of the glucopyranosyl moiety was determined at C-3α (Figure 1). Based on a previous publication, 6 had good compatibility with (+)-lyoniresinol 3α-O-β-D-glucopyranoside and (−)-lyoniresinol 3α-O-β-D-glucopyranoside.16 Therefore, 6 was identified as a mixture of (±)-lyoniresinol 3α-O--D-glucopyranoside.
Five compounds, 1, 2, 4, 5, and 6, were evaluated for their α-glucosidase inhibitory capacity. The activities of compounds 2 (IC = 202.40 M), 4 (182.87 M), and 5 (139.51 M) were better than that of the positive control, acarbose (209.80 M), while compound 1 was weaker than acarbose and mixture 6 was inactive for α-glucosidase inhibition.
Caffeine (1) is the main alkaloid in coffee. According to Herawati's research, caffeine is not a key bioactive component for antioxidant, anti-α-glucosidase, and antiglycation activities.17 Phenolic acids, specifically 5-O-caffeoylquinic acid, 4-O-caffeoylquinic acid, and 3-O-caffeoylquinic acid, are major contributors to antioxidant activity as well as prospective inhibitors against α-glucosidase. The paper by Duangjai also mentioned caffeine's ineffective glucosidase inhibition but highly potent amylase inhibitory activity.18 The IC value of compound 1 in our investigation was higher than that of acarbose. It makes sense that caffeine has a limited anti-α-glucosidase activity.
In the Phoopha report, uridine (2) had no effect on α-glucosidase inhibition.19 The IC value of compound 2 was the lowest among those of the isolated compounds in our study.
Kelampayoside A (4) has been evaluated at 1.0 × 10 M and found to be inactive for human tumor cytotoxicity, anti-inflammatory activity, antioxidant activity, anti-HIV activity, and neuroprotective activity, but it has inhibitory activity against protein tyrosine phosphatase 1B.20 To the best of our knowledge, there has not yet been any report on an α-glucosidase inhibitory assay applied to kelampayoside A (4). Our investigation found that compound 4 (IC= 182.87 µM) inhibited α-glucosidase better than acarbose (IC= 209.80 µM).
Thao reported that the sucrase inhibitory experiment revealed a considerable α-glucosidase inhibitory activity for polystachyol (5).21 In our investigation utilizing a colorimetric reaction, compound 5 (IC= 139.51 µM) also exhibited α-glucosidase inhibitory activity better than acarbose (IC= 209.80 µM).
Research from Jong-Anurakkun revealed that (-)-lyoniresinol 3α-O--D-glucopyranoside exhibited inhibitory activity against both sucrase and maltase with IC values of 1.95 and 1.43 mM, respectively, while its enantiomer was inactive.22 In our investigation, a mixture of (+)-lyoniresinol 3α-O--D-glucopyranoside and (-)-lyoniresinol 3α-O--D-glucopyranoside (6) was isolated and was unable to separate. It was shown to be inactive against α-glucosidase inhibition.
CONCLUSION
From the n-hexane and ethyl acetate extract of Coffea canephora trunk, five compounds and a mixture were isolated, including two alkaloid (caffeine and uridine), a phenolic compound (3,4,5-trimethoxyphenol) and three phenolic glycosides (kelampayoside A, polystachyol, and (±)-lyoniresinol 3α-O--D-glucopyranoside). Based on NMR and MS data analysis as well as comparison to published values, their chemical structures were elucidated. To the best of our knowledge, all the isolated compounds were investigated from the genus Coffea for the first time with the exception of 1. Compounds 2, 4 and 5 were discovered to be potent α-glucosidase inhibitors that are stronger than acarbose, a popular commercial medicine.
ABBREVIATIONS
CNMR: Carbon-13 Nuclear Magnetic Resonance
calcd.: calculated
CC: Chromatographic column
COSY: Homonuclear COrrelation Spectroscopy
d: doublet dd:doublet of doublets m: multiplet s: singlet
HMBC: Heteronuclear Multiple Bond Correlation
HSQC: Heteronuclear single quantum coherence
HR-ESI-MS: High Resolution-ElectroSpray Ionization-Mass Spectrometry
HNMR: Proton Nuclear Magnetic Resonance
IC: The half maximal inhibitory concentration
RP: reversed-phase
COMPETING INTEREST
The authors declare no competing financial interest.
AUTHORS’ CONTRIBUTION
Vo Thi Nga and Nguyen Thi Anh Tuyet contributed to designing the experiments, acquiring the data, interpreting the data and giving final approval of the manuscript to be submitted. Hoang Thi Hien, Nguyen Vu Nhat Ha, Le Thi Minh Phuong, Nguyen Thi Thucarried out the experiments, interpreted NMR and MS data and searched the bibliography.
Corresponding author: Dr. Nguyen Thi Anh Tuyet, Department of Chemistry, Ho Chi Minh City University of Education, Ho Chi Minh City. Email: tuyetnta@hcmue.edu.vn
ACKNOWLEDGMENTS
Thanks are extended to Ho Chi Minh City University of Technology and Education, who supported the facilities for this research.