

Flavonoid C-glucosides and monosaccharides from Combretum indicum
- Sai Gon University, Ho Chi Minh City, Viet Nam
- University of Medicine and Pharmacy at Ho Chi Minh City, Ho Chi Minh City, Viet Nam
Abstract
Introduction: Combretum indicum belongs to Combretaceae. This species was known to possess diverse bioactivities and chemical constituents. This paper reported the isolation and structural elucidation of five compounds from leaves of Combretum indicum collected in Ho Chi Minh City.
Method: From the dried powder of Combretum indicum, the crude extract was obtained by maceration method in methanol at room temperature. Then this extract was separated into different extracts by absorbed on silica gel and eluted with different solvents. The organic compounds were then isolated by chromatography methods and were identified by MS and NMR spectra interpretation.
Results: Five compounds comprising vitexin (1), orientin (2), isoorientin (3), D-glucose (4), and D-fructose (5) were isolated.
Conclusion: To the best of our knowledge, all isolated compounds were reported from this species for the first time.
Keywords: Combretum indicum, flavonoid C-glucoside, monosaccharide
INTRODUCTION
Combretum indicum (L.) DeFilipps., also known as Quisqualis indica, belongs to the Combretaceae family and is widely grown as an ornamental around houses or in parks. Different parts of this plant have been used in folk medicine to treat worms, fever, headache, toothache, and rheumatism1. Extracts or isolated compounds from the leaves, roots, flowers, and bark of this species have been biologically evaluated and shown to have potential antibacterial, antifungal, antioxidant, anticancer, acetylcholinesterase inhibition, and pain relief effects2, 3, 4, 5, 6, 7. This species was previously reported to possess flavonoids, phenolics, steroids, terpenoids, aliphatic acids, and alkaloids7, 8, 9, 10. This paper reports the isolation and structural elucidation of three flavonoid C-glycosides and two monosaccharides from the leaves of Combretum indicum collected in Ho Chi Minh City to contribute to the understanding of the chemical constituents of Combretum indicum species.
MATERIALS AND METHODS
General experimental procedures
The NMR and MS spectra were registered at the Vietnam Academy of Science and Technology on Bruker Avance at 500 MHz for H-NMR and 125 MHz for C-NMR and on an X500 QTOF for ESI-MS.
Plant material
Combretum indicum was collected in Ho Chi Minh City, Vietnam, in March 2019. The scientific name of this plant was identified by PhD Dang Van Son, Institute of Tropical Biology. A voucher specimen (N UMP-BS001) was deposited in the Faculty of Basic Sciences, University of Medicine and Pharmacy at Ho Chi Minh City.
Extraction and isolation
Fresh leaves of Combretum indicum were washed, dried, and ground into powder. This powder was macerated in methanol at room temperature to prepare the crude extract (700 g). Then, the crude extract (700 g) was absorbed on silica gel (1.5 kg) and macerated in n-hexane, n-hexane-ethyl acetate (1:1), ethyl acetate, and methanol. The filtrated solutions were evaporated under reduced pressure to obtain the corresponding extracts.
The n-hexane-ethyl acetate extract (166.8 g) was separated into 20 fractions (HE1-HE20) by silica gel column chromatography (CC) and eluted with n-hexane-ethyl acetate (stepwise, 95:5; 9:1; 8:2; 7:3; 5:5, 0:1) and then ethyl acetate-methanol (stepwise, 9:1; 8:2; 5:5, 0:1). Fraction HE18 (6.93 g) was subjected to silica gel CC and eluted with chloroform:methanol (4:1) to give 12 fractions (A1-A12). Then, 52.3 mg of A9 was washed with methanol to afford 1 (45.9 mg). Fraction A11 (2.3 g) was subjected to reversed-phased silica gel CC and eluted with water:methanol (9:1) to give 17 subfractions (A11.1-A11.17). Next, compounds 2 (35.5 mg) and 3 (12.4 mg) were isolated from subfractions A11.9 (41.9 mg) and A11.11 (23.1 mg), respectively, by washing these fractions with methanol. Subfraction A11.2 (0.84 g) was separated into 3 subfractions by reversed-phased silica gel CC and eluted with water. Then, subfraction A11.2.1 was subjected to normal-phased silica gel CC and eluted with a solvent mixture of chloroform:methanol:water (50:9:1) to obtain 4 (15.1 mg) and 5 (9.3 mg) compounds.

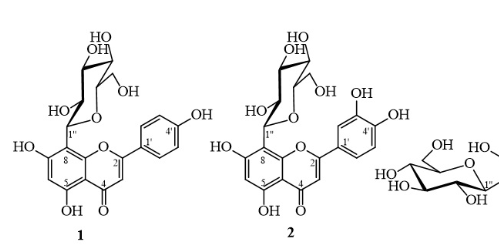
The chemical structures of isolated compounds 1-5
RESULTS
From the n-hexane-ethyl acetate extract of Combretum indicum collected in Ho Chi Minh City, five compounds, 1 (45.9 mg), 2 (35.5 mg), 3 (12.4 mg), 4 (15.1 mg), and 5 (9.3 mg), were isolated. Their physical properties and spectroscopic data were obtained as follows.
Vitexin (1): A yellowish solid. ESI-MS: m/z 431.17 [M‒H]. H-NMR (DMSO-d) (J in Hertz): δ 6.77 (s, H-3), 6.27 (s, H-6), 8.01 (d, 8.0, H-2’/H-6’), 6.89 (d, 8.0, H-3’/H-5’), 4.68 (d, 10.0, H-1’’), 3.84 (t-like, 9.5, H-2’’), 3.24-3.37 (m, H-3’’, H-4’’, H-5’’), 3.75 (brd, 10.5, H-6’’a), 3.53 (dd, 11.5, 5.5, H-6’’b), and 13.16 (s, 5-OH). C-NMR (DMSO-d): δ 163.9 (C-2), 102.4 (C-3), 182.1 (C-4), 160.4 (C-5), 98.2 (C-6), 162.7 (C-7), 104.6 (C-8), 156.0 (C-9), 104.0 (C-10), 121.6 (C-1’), 128.9 (C-2’/C-6’), 115.8 (C-3’/C-5’), 161.1 (C-4’), 73.4 (C-1’’), 70.8 (C-2’’), 78.7 (C-3’’), 70.6 (C-4’’), 81.8 (C-5’’), and 61.3 (C-6’’).
Orientin (2): A yellowish solid. ESI-MS: m/z 446.91 [M‒H]. H-NMR (DMSO-d) (J in Hertz): δ 6.63 (s, H-3), 6.27 (s, H-6), 7.48 (brs, H-2’), 6.87 (d, 8.5, H-5’), 7.52 (brd, 8.0, H-6’), 4.67 (d, 10.0, H-1’’), 3.84 (t-like, 9.5, H-2’’), 3.24-3.27 (m, H-3’’, H-4’’, H-5’’), 3.78 (d, 11.5, H-6’’a), 3.54 (dd, 11.5, 6.5, H-6’’b), and 13.16 (s, 5-OH). C-NMR (DMSO-d): δ 164.1 (C-2), 102.4 (C-3), 182.0 (C-4), 160.4 (C-5), 98.1 (C-6), 162.6 (C-7), 104.5 (C-8), 156.0 (C-9), 104.0 (C-10), 122.0 (C-1’), 114.1 (C-2’), 145.8 (C-3’), 149.6 (C-4’), 115.7 (C-5’), 119.4 (C-6’), 73.4 (C-1’’), 70.8 (C-2’’), 78.8 (C-3’’), 70.7 (C-4’’), 82.0 (C-5’’), and 61.6 (C-6’’).
Isoorientin (3): A yellowish solid. ESI-MS: m/z 447.00 [M‒H]. H-NMR (DMSO-d) (J in Hertz): δ 6.64 (s, H-3), 6.46 (s, H-8), 7.39 (d, 2.0, H-2’), 6.87 (d, 8.0, H-5’), 7.40 (dd, 8.0, 2.0, H-6’), 4.58 (d, 10.0, H-1’’), 4.04 (t-like, 9.5, H-2’’), 3.11-3.22 (m, H-3’’, H-4’’, H-5’’), 3.67 (brd, 10.5, H-6’’a), 3.40 (overlap, H-6’’b), and 13.55 (s, 5-OH). C-NMR (DMSO-d): δ 163.6 (C-2), 102.7 (C-3), 181.8 (C-4), 160.7 (C-5), 108.9 (C-6), 162.8 (C-7), 93.6 (C-8), 156.2 (C-9), 103.2 (C-10), 121.3 (C-1’), 113.2 (C-2’), 145.8 (C-3’), 149.9 (C-4’), 116.0 (C-5’), 119.0 (C-6’), 73.1 (C-1’’), 70.2 (C-2’’), 79.0 (C-3’’), 70.6 (C-4’’), 81.5 (C-5’’), and 61.5 (C-6’’).
D-Glucose (4): A colorless solid. ESI-MS: m/z 179.66 [M‒H]. H-NMR (DO) (J in Hertz) data were obtained for α-D-glucopyranose δ5.28 (d, 4.0, H-1), 3.59 (dd, 9.5, 3.5, H-2), 3.77 (t-like, 9.5, H-3), 3.46 (t-like, 9.5, H-4), 3.88 (m, H-5), 3.91 (m, H-6a), and 3.82 (dd, 14.0, 5.5, H-6b); β-D-glucopyranose δ 4.69 (d, 8.0, H-1), 3.30 (t-like, 9.0, H-2), 3.54 (t-like, 9.0, H-3), 3.45 (t-like, 10.0, H-4), 3.50 (m, H-5), 3.94 (dd, 12.5, 2.0 H-6a), and 3.78 (dd, 12.0, 6.0, H-6b). The C-NMR (CDOD) data of α-D-glucopyranose were δ 94.0 (C-1), 74.0 (C-2), 78.1 (C-3), 73.0 (C-4), 74.9 (C-5), and 62.8 (C-6); those of β-D-glucopyranose were δ 98.2 (C-1), 76.3 (C-2), 78.0 (C-3), 71.9 (C-4), 71.8 (C-5), and 62.9 (C-6).
D-Fructose (5): A colorless solid. ESI-MS: m/z 179.58 [M‒H]. H-NMR (DO) (J in Hertz) data were obtained for β-fructopyranose δ 3.78 (d, 12.0, H-1a), 3.63 (d, 11.5, H-1b), 3.86 (d, 10.0, H-3), 3.96 (dd, 10.0, 3.0, H-4), 4.06 (m, H-5), 4.09 (dd, 12.5, 1.0 H-6a), and 3.77 (d, 13.0, H-6b) and for β-fructofuranose δ 3.64 (d, 12.0, H-1a), 3.62 (d, 12.0, H-1b), 4.17 (m, H-3), 4.16 (m, H-4), 3.89 (m, H-5), 3.87 (d, 12.5 H-6a), and 3.73 (m, H-6b). C-NMR (CDOD) data of β-fructopyranose were obtained; δ 65.9 (C-1), 99.2 (C-2), 69.4 (C-3), 71.8 (C-4), 71.2 (C-5), and 64.5 (C-6); and β-fructofuranose δ 64.5 (C-1), 103.2 (C-2), 77.6 (C-3), 76.8 (C-4), 83.3 (C-5), and 64.2 (C-6).
DISCUSSION
Compound 1 was isolated as a yellowish powder. The H-NMR spectrum of 1 showed signals corresponding to a flavonoid skeleton characterized by a singlet proton at δ 13.16 (1H, s, 5-OH) of a hydroxy group indicating an intramolecular hydrogen bond with a ketone group. A pair of signals with a large coupling constant, each integrated with two protons at δ 8.01 (d, 8.5 Hz, H-2’/H-6’) and δ 6.89 (d, 8.0 Hz, H-3’/H-5’), were assigned to a para-disubstituted benzene ring (ring B). In addition, in the aromatic region of the proton spectrum, two singlet signals were observed at 6.27 (s, H-6) and 6.77 (s, H-3). These results suggested that 1 was an apigenin skeleton. At a higher magnetic field, an anomeric proton signal appeared as a doublet proton signal with a large coupling constant of 10.0 Hz, and a series of carbinol proton signals from 3.24 to 3.84 ppm demonstrated the presence of a β-sugar unit. Its C-NMR spectrum showed 19 carbon signals, consisting of 6 signals corresponding to a hexose skeleton and 13 signals corresponding to an apigenin skeleton. Among these 13 carbon signals, there was one carbonyl carbon at δ 182.1, five oxygenated quaternary carbons in the range from 156.0-163.9 ppm, and two signals at δ 115.8 and 128.9, corresponding to the double intensity of the symmetrical benzene ring of apigenin. The six carbinol carbons of the hexose appeared at 61.3-81.8 ppm, demonstrating that the hexose was attached to apigenin via C-glucoside linkage because the anomeric carbon of a flavone O-glucoside normally resonated at approximately 100 ppm. According to the literature11, 12, 13, 14, in flavone 8-C-glucoside, the methine carbon C-6 resonated at approximately 98-100 ppm, while the methine carbon C-8 of flavone 6-C-glucoside appeared at nearly 93-94 ppm. In the case of compound 1, an aromatic methine carbon resonated at δ 98.2 ppm; therefore, 1 was suggested to be a β-hexose attached to apigenin via an 8-C-glucoside linkage. The ESI-MS spectrum of 1 showed a quasimolecular ion peak at m/z 431.17 [M-H]. Based on the above analysis and the good compatibility of its NMR data with published data 13, 1 was subsequently elucidated to be apigenin 8-C-glucoside (or vitexin).
Compound 2 was isolated as a yellowish powder. Like those of 1, the spectra of 2 displayed signals corresponding to a flavone glycoside. For instance, the proton spectrum revealed a chelated proton signal at δ 13.16 (1H, s) of a hydroxy group at C-5, corresponding to a normal, aromatic protons at 6.27 to 7.52 ppm; an anomeric proton corresponding to a β-sugar at 4.67 (1H, d, 10.0); and a series of carbinol protons at 3.24 to 3.84 ppm. However, 2 exhibited a set of three signals at δ7.52 (1H, brd, 8.0 Hz, H-6’), 7.48 (1H, brs, H-2’), and 6.87 (1H, d, 8.5 Hz, H-5’) for a 1,3,4-trisubstituted benzene ring instead of the para-disubstituted one (ring B), as in 1. These results suggested that 2 possessed a luteolin skeleton. This band corresponded to the presence of 21 carbon signals, including 6 signals corresponding to hexose and 15 signals corresponding to luteolin. The C-NMR spectrum of 2 had 6 carbinol carbons that resonate at 61.6-82.0 ppm from the C-glucoside unit, as in 1. The aromatic methine carbon resonating at δ 98.1 ppm was assigned to C-6, indicating that a sugar unit was attached to luteolin via an 8-C-glucoside linkage. The ESI-MS spectrum of 2 showed a deprotonated ion peak at m/z 446.91 [M-H]. Additionally, upon comparison of the NMR data of 2 with those published in the literature11, 12, 2 was determined to be luteolin 8-C-glucoside (or orientin).
Compound 3 was isolated as a yellowish powder. The NMR spectra of 3 displayed many similarities to those of 2, including the number of signals, integration, coupling constant, and resonating region of the signals. However, they had slight differences in chemical shift values, suggesting that they differed from each other in terms of their position of substitution. In detail, the C-NMR spectrum of 3 showed an aromatic methine carbon at δ 93.6, which was assigned to C-8 according to the above analysis; therefore, 3 should be a 6-C-glucoside. The ESI-MS spectrum of 3 showed a pseudomolecular ion peak at m/z 447.00 [M-H]. Therefore, 3 was determined to be luteolin 6-C-glucoside (or isoorientin) because of the good compatibility of its NMR data with previous data 11, 13.
Compound 4 was isolated as a colorless solid. The proton spectrum of 4 showed signals resonating from 3.2 to 5.3 ppm, including two anomeric protons at δ 5.10 (1H, d, 4.0 Hz, H-α) and 4.46 (1H, d, 7.5 Hz, H-β) with a ratio of 1:1.8. Therefore, 4 could be a reducing sugar that exists simultaneously in a mixture of α and β forms. Moreover, the C-NMR spectrum revealed 12 carbon signals in which two anomeric carbon signals resonated at δ 93.8 (C-1α) and 98.2 (C-1β), and ten carbinol carbons resonated from 62.8 to 78.1 ppm. These results suggested that 4 was a hexose-aldose. The analysis of the coupling constants of protons H-2, H-3, and H-4 for both the α and β forms demonstrated their axial positions because of the large coupling constants of approximately 8.0 to 10.0 Hz. Additionally, the ESI-MS spectrum of 4 showed a pseudomolecular ion peak at m/z 179.66 [M-H]. Based on the aforementioned analysis, 4 was determined to be D-glucopyranose by a good fit of its proton data with D-glucopyranose in the same solvent (DO)15 and its C-NMR data16.
Compound 5 was isolated as a colorless solid. Like those of 4, the NMR spectra of 5 showed signals corresponding to a monosaccharide. The C-NMR spectrum showed 12 carbon signals, including two anomeric carbons at δ 99.2 and 103.2, appearing at different intensities. These two anomeric carbons were determined to be quaternary by the absence of correlation in the HSQC spectrum. Therefore, 5 could be a hexose-ketose. Analysis of the set of highly intense proton signals revealed two pairs of methylene protons at δ 4.01 (1H, dd, 12.5, 1.5 Hz, H-6a) and 3.64 (1H, dd, 12.0, 2.0 Hz, H-6b); 3.66 (1H, d, 11.0 Hz, H-1a) and 3.49 (1H, d, 11.0 Hz, H-1b) which had HSQC correlations with carbons at δ 64.5 (C-6) and 65.9 (C-1), respectively, and showed HMBC correlations with the anomeric carbon at 99.2 (C-2). Moreover, the presence of carbinol protons at δ 3.78 (1H, d, 9.3 Hz, H-3) and 3.79 (1H, dd, 9.5, 3.0 Hz, H-4) confirmed the axial positions of H-3 and H-4. These signals are characteristic of β-fructopyranose. A set of signals with lower intensities was assigned for β-fructofuranose, as shown in the literature17, 18. All proton and carbon signals were confirmed by HSQC and HMBC correlations. The APCI-MS spectrum of 5 showed a deprotonated ion peak at m/z 179.58 [M-H]. Based on the above information and the good compatibility of its NMR data with those published in the literature17, 18, 5 was elucidated as D-fructose.
CONCLUSION
From the n-hexane-ethyl acetate extract of Combretum indicum, five compounds were isolated, which consisted of three flavonoid C-glucosides (vitexin, orientin, and isoorientin) and two monosaccharides (D-glucose and D-fructose). Their chemical structures were elucidated by NMR and HR-MS data analysis, along with the comparison of these data to published data. The presence of all five isolated compounds in Combretum indicum was reported for the first time.
ABBREVIATIONS
ESI-MS: Electrospray ionization-mass spectrometry
APCI-MS: Atmospheric pressure chemical ionization-Mass spectrometry
H-NMR: Proton nuclear magnetic resonance
C-NMR: Carbon-13 nuclear magnetic resonance
HSQC: Heteronuclear single quantum coherence
HMBC: Heteronuclear multiple bond correlation
s: singlet
d: doublet
dd:doublet of doublets
t: triplet
m: multiplet
DMSO: dimethyl sulfoxide
ACKNOWLEDGEMENT
This research was supported by the University of Medicine and Pharmacy at Ho Chi Minh City, grant number 3751/QÐ-ÐHYD.
COMPETING INTEREST
The authors declare no competing financial interest.
AUTHORS’ CONTRIBUTION
Nguyen T.D., Pham T.H.H., and Truong T.T.D. contributed to conducting the experiments and acquiring the data. Pham N.K.T. and Nguyen T.H.T. interpreted the NMR and MS data and gave final approval for the manuscript to be submitted. All authors read and approved the final manuscript.