

Studying the inhibition mechanism of human acetyl-CoA carboxylase by aromatic ligands in the treatment of fatty acid metabolic syndrome using computational chemistry methods
- Chemistry, Physical chemistry, Ho Chi Minh city, University of Natural Science, Nguyen Van Cu, Nguyen Cu Trinh subdistrict, District 1, Ho Chi Minh city, Ho Chi Minh, 700000, Ho Chi Minh, Viet Nam
- Medicine, Biochemistry, Ho Chi Minh City, University of Medicine and Pharmacy, 217 Hong Bang, Ho Chi Minh, 700000, Ho Chi Minh, Viet Nam
Abstract
Background: Metabolic syndrome is a consequence of excess fatty acid accumulation in the human body, causing metabolic disorders related to the role of the two acetyl-CoA carboxylase (ACC) isoforms, ACC1 and ACC2. Inheriting the results from our previous research about "the effective model of human acetyl-CoA carboxylase inhibition by aromatic-structure inhibitors", this research explains the inhibition mechanism of the enzyme by studying the interaction direction, preferential binding sites of aromatic ligands on the isoforms and conformational changes of enzyme-ligand complexes in the body’s environmental conditions (temperature 37 degrees Celsius and water solvent).
Methods: This research applies hard docking combined with molecular dynamics and virtual screening of 14 inhibitory ligand groups, which have 50% enzyme inhibitor (IC50) values determined by experiments, including bipiperidylcarboxamide derivatives, polyketides, aryloxyphenoxypropionate, cyclohexanedione, synthetic acyl-CoA fatty acid chains, acylsulfonamide derivatives, Taisho inhibitors, Sanofi-Aventis inhibitors, AstraZeneca inhibitors, Takeda inhibitors, Pfizer inhibitors, Nimbus inhibitors, Amgen inhibitors and Boehringer Ingel-heim inhibitors. The geometric structures of the ligands are modeled and optimized by quantum mechanical methods, including PM3 for ACC1 and DFT at the B3LYP/6-31G(d,p) level for ACC2 based on our previous research about the QSAR enzyme-inhibition models, using Gaussian 09 W software.
Results: Binding energy values at the identified preferential binding sites of aromatic ligands are closely correlated with the experimental log(IC50), which has statistical significance (p < 0.05): R2ACC1 = 96.3% and R2ACC2 = 78.9%. Those sites reveal the five reaction niches on ACC1, involving the two preferential binding regions in the order of amino acids 152 to 643 and 1632 to 2100, and the four reaction niches on ACC2, involving the two preferential binding regions in the order of amino acids 270 to 704 and 1831 to 2299. The molecular dynamic simulation of enzyme-ligand complexes in the body’s environmental conditions confirms that if ligands have fewer stereogenic interactions and are ready to form four types of bonds, including hydrogen, pi-alkyl, pi-cation and alkyl-alkyl bonds, their inhibitory activity will be higher.
Conclusion: The research clarifies the inhibition mechanism of the enzyme in which ligands prefer to interact with amino acids 152 to 643 and 270 to 704 on the biotin carboxylase domain of ACC and amino acids 1632 to 2100 and 1831 to 2299 on the carboxyl transferase domain of ACC. In addition, the fewer stereogenic interactions of ligands and the easier it is to form four types of bonds, including hydrogen, pi-alkyl, pi-cation and alkyl-alkyl bonds, the higher their inhibitory activity.
Introduction
ACC is an enzyme contributing to the regulation of fatty acid biosynthesis and oxidation and consists of two isoforms, ACC1 and ACC21. It is also an important mediator of carbohydrate, amino acid and lipid metabolism. Abnormalities in metabolism related to the roles of ACC1 and ACC2 lead to excess fatty acids in the body and cause metabolic syndrome (MetS)2.
MetS is a series of cardiovascular risk factors, including abdominal fat, glucose intolerance, triglyceride increment and hypertension, related to an increment in crude fat content in the human body. This leads to a higher chance of stroke, type 2 diabetes and cardiac death. According to the National Health and Nutrition Examination Survey (NHANES), during the past decade, the proportion of men and women aged between 12 and 19 with MetS has increased by more than 50%. In addition, there are approximately more than 200 million adolescents worldwide who have suffered from MetS today2. In supporting the treatment of fatty acid metabolic disorder at the molecular level, the inhibition of ACC activity plays an important role in constraining the synthetic biology reaction rate of fatty acids (ACC1) and promoting their oxidation process (ACC2) in the human body3.
Many studies have demonstrated that ACC inhibition offers potential in controlling fatty acid biosynthesis, leading to the development of new solutions for the prevention and treatment of MetS-related diseases. Studies by Jeffrey and Matthew (2014) reported an ACC inhibitor model performed on yeast4. Accordingly, these inhibitors have diverse and complex structures, including bipiperidylcar-boxamide derivatives, polyketides (soraphen A), aryloxyphenoxypropionate (haloxyfop), cyclohex-anedione (sethoxydim), synthetic acyl-CoA fatty acid chains (TOFA, MEDICA), competitive substrate inhibitors (CABI-CoA), acylsulfonamides and related derivatives. In addition to the above inhibitor groups, many studies by Taisho, Sanofi-Aventis, AstraZeneca, Takeda, Pfizer, Boehringer Ingelheim, Haselkorn, Nimbus and Amgen, which were performed on yeast and mice, resulted in several other nonselective inhibitor structures that can be attached to both the carboxyl transferase (CT) and biotin carboxylase (BC) active sites at the same time.
Inheriting the results from our previous research about "the effective model of human acetyl-CoA carboxylase inhibition by aromatic-structure inhibitors"5, the aim of this research is to reveal the inhibition mechanism of the enzyme by studying the interaction direction, preferential binding sites of aromatic ligands on the two isoforms and conformational changes of enzyme-ligand complexes in the body’s environmental conditions (temperature 37 degrees Celsius and water solvent).
Software and Methods
Modeling and optimizing ligands
Gaussian 09w6 is utilized to model and optimize the 14 inhibitory ligand groups, including 21 inhibitory ligands on ACC1 (
21 inhibitory ligands on ACC1
|
Ligands |
Log(IC50) |
|---|---|
|
Taisho1 |
2.00 |
|
Taisho2 |
1.52 |
|
Taisho3 |
1.87 |
|
Taisho4 |
1.38 |
|
Taisho5 |
2.28 |
|
Sanofi2 |
2.28 |
|
Astra5 |
4.11 |
|
Astra6 |
3.08 |
|
Astra7 |
3.00 |
|
Takeda1 |
2.74 |
|
Takeda2 |
1.50 |
|
Takeda3 |
2.08 |
|
Takeda4 |
2.20 |
|
Takeda5 |
2.11 |
|
Takeda6 |
1.32 |
|
Pfizer4 |
3.40 |
|
Pfizer5 |
2.07 |
|
Pfizer6 |
1.00 |
|
Nimbus1 |
1.00 |
|
Amgen1 |
3.66 |
|
Amgen2 |
2.28 |
29 inhibitory ligands on ACC2
|
Ligands |
Log(IC50) |
|---|---|
|
Taisho1 |
1.36 |
|
Taisho2 |
1.52 |
|
Taisho3 |
1.87 |
|
Taisho4 |
1.38 |
|
Taisho5 |
1.98 |
|
Sanofi1 |
2.80 |
|
Sanofi2 |
1.48 |
|
Astra1 |
3.40 |
|
Astra2 |
3.56 |
|
Astra3 |
2.78 |
|
Astra4 |
2.32 |
|
Astra5 |
3.11 |
|
Astra6 |
2.04 |
|
Astra7 |
2.00 |
|
Takeda1 |
1.53 |
|
Takeda2 |
0.73 |
|
Takeda3 |
1.26 |
|
Takeda4 |
0.93 |
|
Takeda5 |
1.36 |
|
Takeda6 |
0.69 |
|
Pfizer3 |
1.04 |
|
Pfizer4 |
3.40 |
|
Pfizer5 |
1.88 |
|
Pfizer6 |
0.60 |
|
Boe1 |
1.70 |
|
Boe2 |
2.26 |
|
Nimbus1 |
1.00 |
|
Amgen1 |
1.57 |
Identification of binding sites on ACC1 and ACC2
The two isoforms are structurally extracted from the RCSB protein data bank7: 2YL2 (BC), 4ASI (CT) for ACC1 (Figure 1) and 3JRX (BC), 3FF6 (CT) for ACC2 (Figure 2).

The model of ACC1 (2YL2 structure) with amino acid sequence 78-617 (Figure A) and (4ASI structure) with amino acid sequence 1493-2260 (Figure B).

The model of ACC2 (3JRX structure) with amino acid sequence 217-775 (Figure A) and (3FF6 structure) with amino acid sequence 1693-2450 (Figure B).
To identify binding sites on ACC1, we utilized Discovery Studio Visualizer 2017 R2 client software8 for 32 ligand binding sites (Figure 3). Accordingly, combined with docking and virtual screening of ligands corresponding to 4 different methods by AutoDock9 and AutoDock Vina 10, including simulated annealing, genetic algorithm, Lamarckian genetic algorithm and hybrid-scoring function, another 12 ligand binding sites were detected.

Binding sites, described as red spheres, on ACC1 determined by Discovery Studio.
In the case of ACC2, 13 ligand binding sites were detected, including 5 binding sites available from X-ray diffraction data (Figure 4) and 8 binding sites from virtual screening.

Binding sites between the soraphen A (described as blue dotted circles) and CP640186 (described as green dotted circles) ligands and the amino acids on BC and CT of ACC2.
Raccoon9 is utilized to run virtual screening of ligand structures by receiving parameters from AutoDock. The docking grid volume is determined by two methods: preselected ligand binding sites identified by Discovery Studio and the grid overlaid with the entire enzyme structure.
Cygwin was utilized to run virtual screening of ligand structures by receiving parameters from Autodock vina, including the enzyme.pdbqt, the ligand.pdbqt and the docking grid volume determined on each preselected ligand binding site identified by Discovery Studio.
Determination of preferential binding sites of ligands on the enzymes
Based on the binding sites identified in the previous step, we utilized R-console software 3.4.211 for infinite loop calculation with stopping conditions (correlation coefficients, r>0.85) and searched for the sites for which the binding energy was most closely correlated with the experimental log(IC). Accordingly, Discovery Studio was utilized to simulate complex structures and determine the types of bonds between the enzymes and ligands.
Evaluating conformational changes of enzyme-ligand complexes in the body’s environmental conditions
To evaluate the conformational changes of enzyme-ligand complexes, we applied the molecular dynamics technique with the TIP3P water-solvent model utilizing Chimera 1.12 software12 in two cases. In the first case, the enzyme-ligand structures had the highest (ACC1-Pfizer6, ACC2-Pfizer6) and the lowest inhibitory activity (ACC1-Astra5, ACC2-Astra1). The second one, ACC2, binds to CP640186 (CP640186_A, CP640186_B, CP640186_C, CP640186_D) and soraphen A. Accordingly, in all cases, we utilized Discovery Studio to analyze the binding patterns of the enzyme average conformations with the lowest potential energy.
Results
QSAR enzyme-inhibition models
The results of our previous research show that in the case of ACC1, the hybrid neural fuzzy inference system (HYFIS) model (Figure 5) of the compounds optimized by the PM3 method indicates that four descriptors, GATS2c, JGI5, ETA_EtaP, and CIC5, correlate closely with logIC (R2 = 84.1%). The higher the ACC1 inhibitory activity is, the higher the number of aromatic rings and the higher the number of atoms belonging to the small period, which have more electronegativity and electron resonance effects, such as O atoms. In the case of ACC2, the HYFIS model (Figure 6) of the compounds optimized by the DFT method indicates that the four descriptors, GATS6s, GATS1e, MATS3s, and GATS3s, correlate closely with logIC (R2 = 90.8%). The higher the ACC2 inhibitory activity is, the more C=O or C=N double bonds, the higher the number of atoms that have the large Sander- son electronegativity (O>N>S) and the small Sanderson electronegativity difference between first order interleaved atoms (the preferable N-X-H structure due to O-X-H > N-X-H).

Membership functions of HYFIS for the case of ACC1 – PM3 optimization.

Membership functions of HYFIS for the case of ACC2 – DFT optimization.
Locating binding sites on the two enzymes
In the case of ACC1, 44 binding sites and binding energy values (BE - kcal/mol) were identified. In the case of ACC2, 13 binding sites and binding energy values were identified.
Correlation between binding energies at preferential binding sites and the experimental log(IC) of ligands on ACC1 and ACC2
For both ACC1 and ACC2, the BE values of ligands are closely correlated with the experimental log(IC), which has statistical significance (p < 0.05): R2ACC1 = 96.3% (Figure 7) and R2ACC2 = 78.9% (Figure 8).
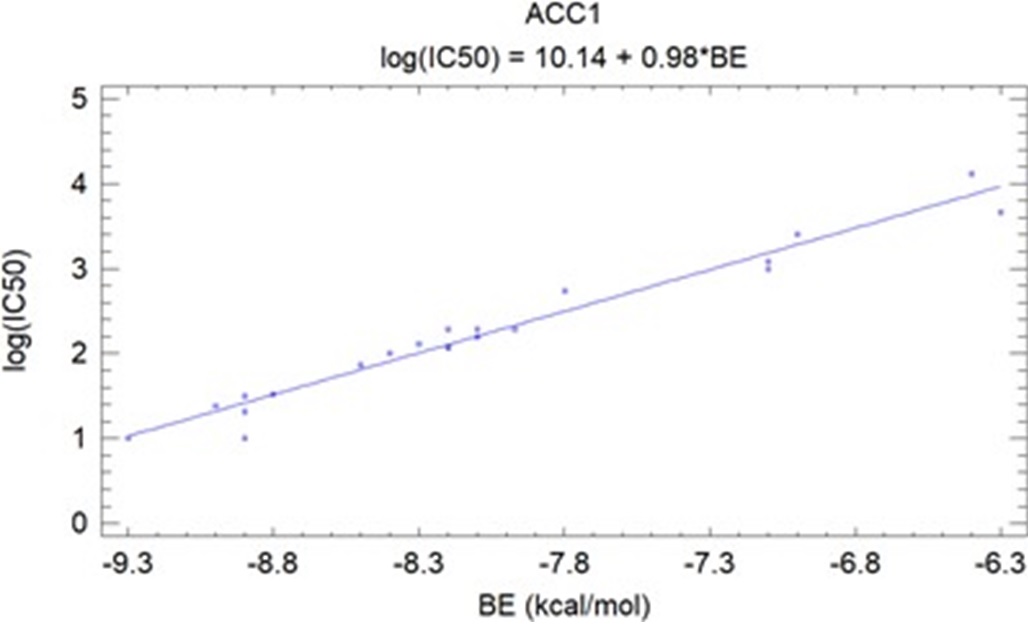
Correlation between binding energy (BE) and the experimental log(IC50) of ligands on ACC1.

Correlation between binding energy (BE) and the experimental log(IC50) of ligands on ACC2.
Investigation of enzyme-ligand complexes in the body’s environmental conditions
The binding patterns of the enzyme average conformations, including the enzyme-ligand complexes associated with Pfizer6 having the highest inhibitory activity and Astra5 — Astra1 having the lowest inhibitory activity on ACC1 (Figure 9) and ACC2 (Figure 10), were analyzed by Discovery Studio.

The average binding patterns of the ACC1-Pfizer6 complex (BE=-9.30) and ACC1-Astra5 complex (BE=-6.40) in the body environment.
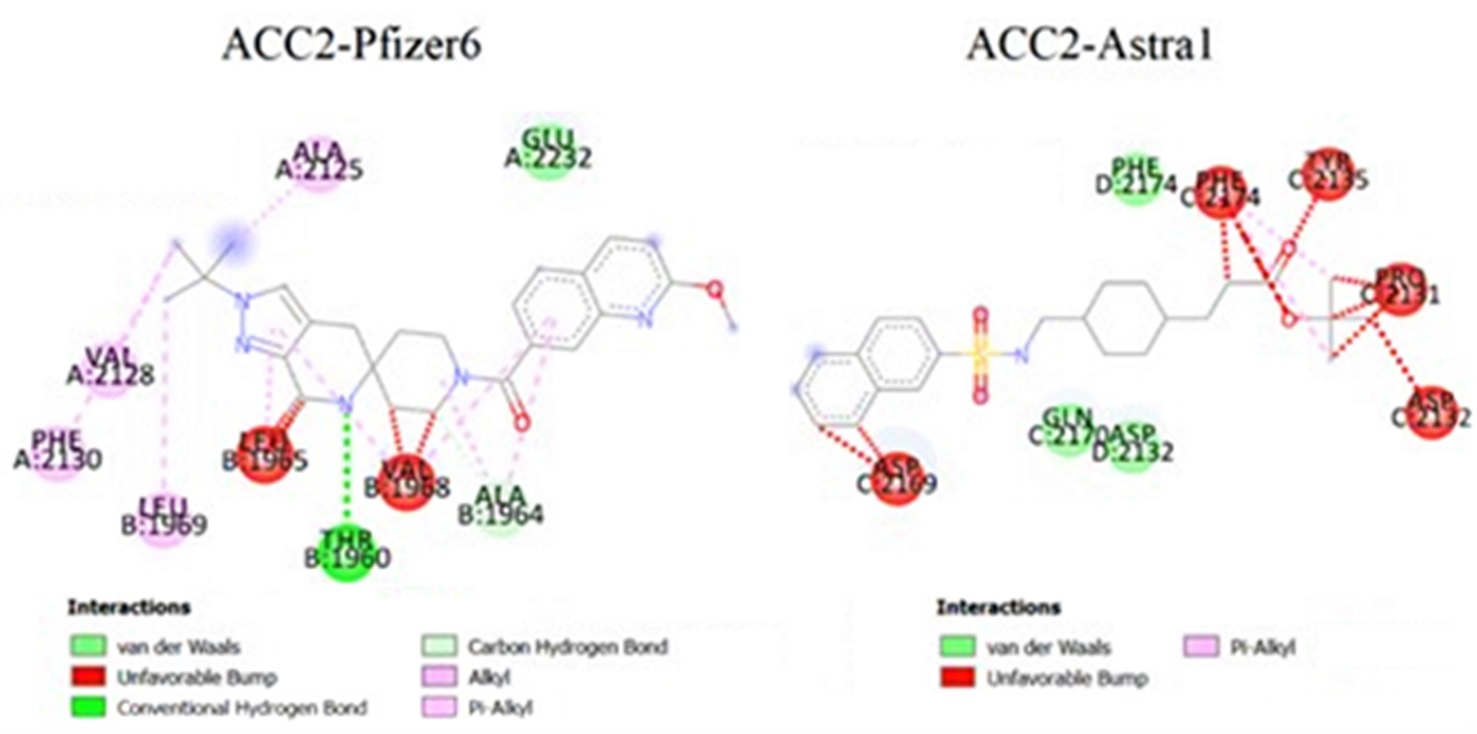
The average binding patterns of the ACC2-Pfizer6 complex (BE=-10.90) and ACC2-Astra1 complex (BE=-7.27) in the body environment.
The preferential binding regions on ACC1 and ACC2
The preferential binding sites of ligands reveal the five reaction niches on ACC1 (Figure 11), involving the two preferential binding regions in the order of amino acids 152 to 643 and 1632 to 2100 (Figure 13), and the four reaction niches on ACC2 (Figure 12), involving the two preferential binding regions in the order of amino acids 270 to 704 and 1831 to 2299 (Figure 14).

The five reaction niches (A – E) on the BC domain and CT domain of ACC1.

The four reaction niches (F – I) on the BC domain and CT domain of ACC2.

The preferential binding regions in the order of amino acids 152 to 643 and 1632 to 2100 on ACC1.

The preferential binding regions in the order of amino acids: 270 to 704 and 1831 to 2299 on ACC2.
Discussion
The suppression of ACC activity is crucial in limiting the synthetic biology reaction rate of fatty acids (ACC1) and encouraging their oxidation process (ACC2) in the human body, which supports therapy of fatty acid metabolic disorder at the molecular level. Numerous studies have shown that ACC inhibition has the capacity to regulate fatty acid production, opening up new avenues for the prevention and treatment of MetS-related disorders. Studies of Cho. (2008) revealed the crystal structure of the BC and CT domains of human ACC213. Timothy's research (2014) discovered the domain crystal structure of some microorganism ACCs14. Jeffrey and Matthew (2014) reported an ACC inhibitor model performed on yeast that resulted in several nonselective inhibitor structures that can be attached to both the carboxyl transferase and biotin carboxylase active site at the same time4. In addition, Chen (2019) reviewed and updated some human ACC inhibitors in clinical studies3. All of these studies have focused on studying structures and ACC inhibition models in yeasts and microorganisms or determining the experimental activity of human ACC inhibitors based on clinical studies. Therefore, the inhibition mechanism of human ACC has not yet been elucidated.
The findings of this study indicate that the inhibition mechanism of the human ACC is explained through the preferential binding sites of ligands on the two isoforms and conformational changes of enzyme-ligand complexes in the body’s environmental conditions. Accordingly, ligands prefer to interact with amino acids 152 to 643 and 270 to 704 on the biotin carboxylase domain and amino acids 1632 to 2100 and 1831 to 2299 on the carboxyl transferase domain of the human ACC, described as the five reaction niches (A – E) on ACC1 and the four reaction niches (F – I) on ACC2. These calculation reaction sites are consistent with the research results of Cho. (2008) about the crystal structure of the BC and CT domain of the human ACC2. In addition, there is a close correlation between the experimental log(IC), based on the research of Matthew (2014) and Chen (2019), and the calculated binding energy values of ligands with statistical significance (p < 0.05). The conformational changes of enzyme-ligand complexes in the body’s environmental conditions, as the new finding of this study, show that each enzyme-ligand complex has an unfavorable bump and breaks three types of unstable bonds: Pi-Pi bonds between the conjugated Pi system of amino acids on the enzyme and the conjugated Pi system of aromatic rings on the ligand, sulfur-Pi bonds between the conjugated Pi system of amino acids on the enzyme and sulfur on the ligand, and Pi-amide bonds between amide groups of amino acids on the enzyme and the conjugated Pi system of aromatic rings on the ligand. In addition, four new types of bonds are formed: hydrogen bonds, Pi-alkyl bonds between the alkyl group of amino acids on the enzyme and the conjugated Pi system of aromatic rings on the ligand; Pi-cation bonds between the cation of amino acids on the enzyme and the conjugated Pi system of aromatic rings on the ligand; and alkyl-alkyl bonds. Therefore, ligands have fewer stereogenic interactions, and the easier it is to form these types of bonds, the higher their inhibitory activity. However, the mechanism of bond breaking and formation has not been clarified and will be reviewed in our future research.
Conclusions
The results of the study have solved the main objectives, including the interaction direction, preferential binding sites of aromatic ligands and conformational changes of enzyme-ligand complexes in the body’s environmental conditions.
The research reveals the five reaction niches on ACC1 and the four reaction niches on ACC2, including the two preferential binding regions in the order of amino acids: 152 to 643 and 1632 to 2100 for ACC1, 270 to 704 and 1831 to 2299 for ACC2.
The results of molecular dynamic simulation of enzyme-ligand complexes in the body’s environmental conditions show that each complex has an unfavorable bump and breaks three types of unstable bonds, including Pi-Pi, sulfur-Pi and Pi-amide bonds. In addition, each complex forms four new types of bonds: hydrogen, pi-alkyl, pi-cation and alkyl-alkyl bonds. If ligands have fewer stereogenic interactions and are ready to form four types of bonds, including hydrogen, pi-alkyl, pi-cation and alkyl-alkyl bonds, their inhibitory activity will be higher.
Abbreviations
ACC: acetyl-CoA carboxylase; BC: biotin carboxylase; CT: carboxyl transferase; MetS: metabolic syndrome; QSAR: quantitative structure-activity relationship; HYFIS: hybrid neural fuzzy inference system; BE: binding energy; IC: half maximal inhibitory concentration.
Competing interests
The authors declare that they have no competing interests.
Funding
This research was funded by Ho Chi Minh City, University of Science.
Authors’ contributions
CMNT carried out calculations and data analysis. TTB conceived of the study and participated in research coordination. TLX provided theories of the research. XTTL drafted the manuscript and designed and supported the research. The authors read and approve the manuscript.
Acknowledgments
None.