

Impacts of basis sets, solvent models, and NMR methods on the accuracy of 1H and 13C chemical shift calculations for biaryls: a DFT study
- Institute of Research and Development, Duy Tan University, Da Nang 550000, Vietnam
- Faculty of Natural Science, Duy Tan University, Da Nang 550000, Vietnam
Abstract
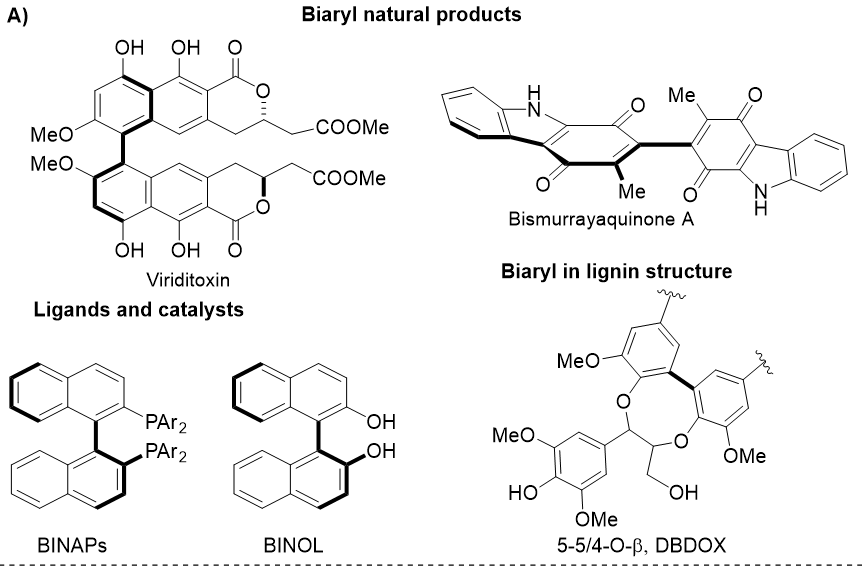
Introduction: Biaryls are core structures composed of chiral ligands, organocatalysts, biologically active natural products and biopolymer lignins. In this study, the effects of basis sets, solvent models, and NMR methods on the accuracy of 1H/13C NMR chemical shift calculations for biaryl structures were evaluated.
Methods: All calculations were performed using Gaussian09. The GIAO NMR results were observed and extracted using GaussView05. To reduce the systematic error of the calculations, linear regression analysis of the calculated chemical shifts versus the experimental shifts was performed.
Results: The tested basis sets showed good 1H/13C results, with CMAE values as low as 0.0425 ppm and 1.09 ppm for 1H and 13C, respectively. The use of solvent models significantly increased the accuracy of the 1H chemical shift calculations. The GIAO method produced more accurate results than did the IGAIM and CSGT methods.
Conclusion: This study recommends 6-31G(d,p) and DGDZVP basis sets, IEMPCM and CPCM solvent models, and GIAO NMR methods for the accurate prediction of 1H and 13C chemical shifts for biaryls, assisting in their full structural assignments.
Introduction
Biaryls are important core structures present in useful chiral ligands, organocatalysts, biologically active natural products, and biopolymer lignins.1, 2 Typical examples illustrated in Figure 1A are -symmetric binaphthyls (BINOL and BINAPs), which catalyze numerous asymmetric transformations3, 4 the mycotoxin viriditoxin 5 the alkaloid bismurrayaquinone A6 and 5-5/4-O- lignin substructures7. These compounds also possess biaryl linkages, which can give rise to atropisomers that have received significant attention from the synthetic community in the last decade.1, 2 An accurate method for predicting NMR spectra would contribute valuable insights into the conformations of biaryl structures and the local electron environment of each NMR active nucleus. Gauge-independent atomic orbital (GIAO)-DFT NMR calculations have effectively supported the structural assignment and validation of biaryl compounds with accurate predictions at affordable computational costs. 6, 8 In general, the accuracy is impacted by optimized geometries, density functional methods, basis sets, solvation models, and NMR methods.7, 8, 9 For the two common nuclei of organic molecules, H shift predictions are more challenging than C shift predictions due to the significant impact of solvation effects on protons.
Previous studies reported how the use of different density functional methods and basis sets for NMR calculations affected the H/C results for a variety of different organic structures. 10, 11, 12 In 2015, Toomsalu reported the use of 18 DFT functionals and 6 basis sets for H and C calculations of small organic molecules and reported that the best functional/basis set for C was PBE1PBE/aug-cc-pVDZ, and those for H were HSEH1PBE, mPW1PW91, PBE1PBE, CAM-B3LYP, and B3PW91 functionals and cc-pVTZ for H. In 2017, Iron recommended LC-TPSSTPSS/cc-pVTZ among an extensive list of tested functionals and basis sets for C predictions. In our continuing interest in the NMR modeling of biaryls, we have recently reported the impact of density functional methods on the accuracy of H/C chemical shift calculations for biaryls.5, 9 Herein, the present study shows how basis sets, solvent models, and NMR methods influence the accuracy of H and C NMR shift calculations for biaryl 1 (Figure 1B).
Computational methods
All calculations were performed using Gaussian0913 on a commercial computer with an Intel Core i3-7100 processor. Geometry optimizations were performed at CAMB3LYP/6-31G(d,p) with default convergence criteria. The integral equation formalism variant of the polarized continuum model (IEFPCM) was incorporated during geometry optimization.14 Subsequent frequency calculations ensured that a potential energy surface (PES) local minimum was attained during energy minimization.

A) Biaryls in natural products, ligands, catalysts, and lignins; and B) Biaryl 1 with numbering labels and its optimized geometry at the level IEFPCM(DMSO)/CAM-B3LYP/6-31G(d,p) of theory.
The following basis sets, solvent models, and NMR methods, which are commonly used for determining H/C NMR chemical shifts, were evaluated:
Unless specified otherwise, single-point NMR GIAO calculations were carried out at the IEFPCM (DMSO)/B97XD/6-31G(d,p) level of theory, which was found to produce computed 1H/13C chemical shifts with high accuracy. The GIAO NMR results were observed and extracted using GaussView05. Each optimized structure was used for computing the corresponding isotropic shielding constants (σ). The chemical shifts (δ) were obtained using Equation 1. For both the 1H and 13C NMR calculations, an average of the values of equivalent atoms was assumed. For example, a single proton/carbon signal is observed for the two methoxy groups of dimer 1 due to fast rotations of the biaryl linkage and two methyl groups relative to the NMR measurement time scale. To reduce the systematic error of the calculations, linear regression analysis of the calculated chemical shifts versus the experimental shifts (δ) (Equation 2)) was performed, and the scaled chemical shifts (δ) were computed according to Equation 3. Linear regression was used based on the fitness of the calculated data. As a reference had a negligible impact on the linear regression analysis, fixed values of 197 ppm and 31 ppm were chosen as the TMS shielding constants for 13C and 1H, respectively. The computed results were evaluated using the mean absolute value (│Δδ│/ppm, Equation 4), corrected mean absolute error (CMAE/ppm, Equation 5), corrected root mean squared error (CRMSE/ppm, Equation 6), and Pearson correlation coefficient (r). Smaller values of CMAE and CRMSE indicate smaller errors, and a larger value of r indicates a stronger correlation between the theoretical and experimental data. Error calculations and linear correlations were performed using Microsoft Excel 2013.
Figure 1 shows the numbered dimers used for the proton and carbon atoms in this study. Due to the axial symmetry of biaryl 1, only one side of the structure was labeled. Compound 1 contains phenolic and carboxylic protons, which typically do not appear in the NMR spectra due to rapid exchanges in DMSO- or CDCl. Therefore, these protons were excluded from the calculations in this study. The experimental H and C NMR spectra of 19, 18 were reported.
Results
Impact of basis sets
9 Basis sets, including 6-31G, 6-31G(d,p), 6-31G(3d,p), 6-31G(d,3p), 6-31G+(d,p), 6-31G++(3d,p), 6-311(d,p), cc-pVDZ, and DGDZVP, were coupled with B97XD and the IEFPCM solvent model (DMSO) for the NMR calculations of compound 1, optimized at the IEFPCM(DMSO)/CAM-B3LYP/6-31G(d,p) level of theory. The calculated H/C shifts, statistical parameters, and absolute deviations are shown in

Absolute deviations (ppm) of the 1H/13C chemical shifts calculated using 9 basis sets
|
Exp. |
6-31G |
6-31G(d,p) |
6-31G(3d,p) |
6-31G(d,3p) |
6-31G+(d,p) |
6-31G++(d,p) |
6-311G(d,p) |
cc-pVDZ |
DGDZVP | |
|
H1 |
6.78 |
6.97 |
6.83 |
6.74 |
6.81 |
6.88 |
6.86 |
6.86 |
6.86 |
6.92 |
|
H5 |
6.56 |
6.34 |
6.50 |
6.61 |
6.53 |
6.48 |
6.50 |
6.47 |
6.46 |
6.43 |
|
H7 |
2.74 |
2.60 |
2.67 |
2.64 |
2.67 |
2.69 |
2.69 |
2.66 |
2.70 |
2.42 |
|
H8 |
2.49 |
2.57 |
2.54 |
2.57 |
2.56 |
2.59 |
2.59 |
2.55 |
2.48 |
2.16 |
|
H10 |
3.79 |
3.87 |
3.82 |
3.81 |
3.78 |
3.71 |
3.71 |
3.82 |
3.86 |
3.52 |
|
C1 |
126.83 |
126.73 |
126.14 |
127.41 |
125.67 |
126.18 |
127.36 |
126.53 |
125.72 |
128.06 |
|
C2 |
123.62 |
125.44 |
125.23 |
124.69 |
124.86 |
126.24 |
126.14 |
124.41 |
125.40 |
122.77 |
|
C3 |
142.69 |
141.75 |
143.78 |
144.50 |
144.28 |
141.76 |
141.39 |
144.17 |
143.59 |
142.94 |
|
C4 |
148.59 |
146.80 |
147.92 |
147.19 |
147.89 |
147.81 |
147.84 |
148.34 |
148.59 |
146.86 |
|
C5 |
111.76 |
110.80 |
110.57 |
112.79 |
109.75 |
108.92 |
109.16 |
109.69 |
110.34 |
111.64 |
|
C6 |
131.94 |
131.58 |
132.25 |
129.62 |
132.12 |
133.30 |
132.67 |
133.01 |
133.34 |
131.01 |
|
C7 |
31.11 |
31.93 |
31.94 |
32.00 |
32.15 |
32.46 |
32.65 |
32.12 |
32.41 |
30.18 |
|
C8 |
36.66 |
39.15 |
38.24 |
37.58 |
38.62 |
37.84 |
37.32 |
38.45 |
38.52 |
38.88 |
|
C9 |
174.95 |
177.33 |
174.95 |
175.06 |
175.66 |
176.05 |
175.93 |
174.57 |
174.28 |
176.68 |
|
C10 |
56.82 |
53.43 |
53.88 |
54.02 |
53.87 |
54.34 |
54.50 |
53.61 |
52.71 |
55.93 |
Accuracy evaluation of 1H and 13C chemical shift calculations using 9 basis sets
|
δ(1H) |
δ(13C) | ||||||
|
Entry |
Basis set |
r2 |
CMAE |
CRMSE |
r2 |
CMAE |
CRMSE |
|
1 |
6-31G |
0.9934 |
0.141 |
0.151 |
0.9985 |
1.50 |
1.80 |
|
2 |
6-31G(d,p) |
0.9992 |
0.0519 |
0.0535 |
0.9992 |
1.09 |
1.34 |
|
3 |
6-31G(3d,p) |
0.9988 |
0.0574 |
0.0646 |
0.9990 |
1.29 |
1.50 |
|
4 |
6-31G(d,3p) |
0.9993 |
0.0425 |
0.0491 |
0.9989 |
1.35 |
1.55 |
|
5 |
6-31G+(d,p) |
0.9978 |
0.0837 |
0.0860 |
0.9987 |
1.53 |
1.71 |
|
6 |
6-31G++(d,p) |
0.9983 |
0.0745 |
0.0768 |
0.9989 |
1.39 |
1.59 |
|
7 |
6-311G(d,p) |
0.9986 |
0.0664 |
0.0696 |
0.9990 |
1.24 |
1.52 |
|
8 |
cc-pVDZ |
0.9987 |
0.0591 |
0.0672 |
0.9986 |
1.46 |
1.78 |
|
9 |
DGDZVP |
0.9984 |
0.0568 |
0.0735 |
0.9993 |
1.09 |
1.26 |
Effects of solvent models and NMR methods
NMR calculations with no solvent and with two solvent models, IEFPCM and CPCM, were carried out, and the results are shown in
1H/13C chemical shifts calculated using different solvent models and NMR methods
|
Exp. |
No solvent |
IEFPCM |
CPCM |
GIAO |
IGAIM |
CSGT | |
|
H5 |
6.56 |
6.41 |
6.50 |
6.51 |
6.50 |
6.47 |
6.47 |
|
H1 |
6.78 |
6.93 |
6.83 |
6.83 |
6.83 |
6.73 |
6.73 |
|
H7 |
2.74 |
2.66 |
2.67 |
2.67 |
2.67 |
2.51 |
2.51 |
|
H8 |
2.49 |
2.57 |
2.54 |
2.54 |
2.54 |
2.36 |
2.35 |
|
H10 |
3.79 |
3.79 |
3.82 |
3.82 |
3.82 |
4.30 |
4.30 |
|
C1 |
126.83 |
126.33 |
126.14 |
126.10 |
126.14 |
125.43 |
125.43 |
|
C2 |
123.62 |
125.16 |
125.23 |
125.18 |
125.23 |
123.24 |
123.23 |
|
C3 |
142.69 |
143.98 |
143.78 |
143.75 |
143.78 |
145.97 |
145.96 |
|
C4 |
148.59 |
148.13 |
147.92 |
147.92 |
147.92 |
149.45 |
149.44 |
|
C5 |
111.76 |
111.17 |
110.57 |
110.60 |
110.57 |
112.22 |
112.22 |
|
C6 |
131.94 |
132.38 |
132.25 |
132.27 |
132.25 |
128.94 |
128.93 |
|
C7 |
31.11 |
31.61 |
31.94 |
31.94 |
31.94 |
31.84 |
31.84 |
|
C8 |
36.66 |
38.68 |
38.24 |
38.24 |
38.24 |
37.53 |
37.52 |
|
C9 |
174.95 |
174.24 |
174.95 |
174.97 |
174.95 |
174.96 |
174.94 |
|
C10 |
56.82 |
53.27 |
53.88 |
53.89 |
53.88 |
55.40 |
55.40 |
Accuracy evaluation of 1H and 13C chemical shift calculations using solvent models
|
δ(1H) |
δ(13C) | ||||||
|
Entry |
Solvent model |
r2 |
CMAE |
CRMSE |
r2 |
CMAE |
CRMSE |
|
1 |
No solvent model |
0.9965 |
0.0940 |
0.110 |
0.9990 |
1.16 |
1.50 |
|
2 |
IEFPCM |
0.9992 |
0.0519 |
0.0535 |
0.9992 |
1.09 |
1.34 |
|
3 |
CPCM |
0.9992 |
0.0515 |
0.0531 |
0.9992 |
1.09 |
1.33 |

Mean absolute values (ppm) of 1H/13C calculations using two solvent models.
NMR calculations were performed atthe IEFPCM(DMSO)/B97XD/6-31G(d,p)//IEFPCM(DMSO)CAM-B3LYP/6-31G(d,p) level of theory using three NMR methods,and the results are summarized in
Accuracy evaluation of 1H and 13C chemical shift calculations using NMR methods
|
δ(1H) |
δ(13C) | ||||||
|
Entry |
NMR method |
r2 |
CMAE |
CRMSE |
r2 |
CMAE |
CRMSE |
|
1 |
GIAO |
0.9992 |
0.0519 |
0.0535 |
0.9992 |
1.09 |
1.34 |
|
2 |
IGAIM |
0.9806 |
0.203 |
0.260 |
0.9988 |
1.24 |
1.62 |
|
3 |
CSGT |
0.9800 |
0.206 |
0.264 |
0.9988 |
1.24 |
1.62 |

Mean absolute values (ppm) of 1H/13C calculations using three NMR methods
Discussion
The use of either IEFPCM (Entry 2, CMAE = 0.0519 ppm) or CPCM (Entry 3, CMAE 0.0515 ppm) produced much better H results than no use of a solvent model, while the C results for these three methods yielded similar accuracies. These results could be explained by the fact that the high exposure of protons to solvent molecules is more obvious than that of carbon nuclei, which are well shielded. All protons had relatively close deviations (Figure 3), except for methoxy proton H10, which showed a low error when no solvent model was employed. For the computed C chemical shifts, noticeable deviations of carbons C2, C8, and C10 were consistently observed.
A significantly greater accuracy for H results was obtained using the GIAO method than using the IGAIM and CSGT methods. The relatively low absolute deviations for the H results obtained using GIAO are clearly observed in Figure 3. Compared to the H calculations, the C results were not strongly impacted by the three tested NMR methods. This observation was expected due to the relatively low impact of the solvent environment and molecular interactions on the carbon nuclei. The CMAE values ranged from 1.09 to 1.24. The C results were obtained with high coefficients of determination (0.9988 ≤ ≤ 0.9992). Noticeable deviations were observed for methoxy proton H10 and carbon atoms C3, C6, and C10 (Figure 4).
Overall, the above results relating to the tested basis sets, solvent models, and NMR methods indicated the importance of utilizing specific methods to obtain the desired accuracy of H/C NMR calculations for biaryl 1. High-accuracy results could be expected when applying these methods to compounds having this core biaryl structure.
Conclusion
The influence of 9 common basis sets, solvent models, and NMR methods on the accuracy of H/C chemical shift calculations for biaryl 1 were evaluated. The tested basis sets showed good H/C results, with CMAE values as low as 0.0425 ppm and 1.09 ppm for H and C, respectively. For the solvent models, the results indicated that solvent incorporation was necessary for improving the accuracy of the H chemical shift calculations, while it had little effect on the computed C chemical shifts. This is expected because carbon nuclei are less exposed to solvent molecules than to protons. The GIAO method outperformed the IGAIM and CSGT methods. This study highly recommends 6-31G(d,p) basis sets for the effective production of both H/C with high accuracy and low computational cost, IEMPCM and CPCM solvent models for obtaining good H results, and GIAO methods for NMR calculations. This work will be useful for assisting in the full H and C NMR assignments of similar biaryls. In the near future, NMR calculations for biaryl natural products possessing interesting biological properties will be conducted.
Acknowledgment
This project was supported by the International Foundation for Science (IFS), Stockholm, Sweden, through a Grant No. 3-I-E-6576-1 to TTN.